Statin drug use has steadily increased over the last several decades, due to the widespread belief that cholesterol reduction is an important step in preventing heart disease. It is undeniable that statins are effective: they can decrease serum cholesterol levels from over 300 db/ml to well within the normal range in a matter of weeks. For a person who already has normal cholesterol levels, statins can drive their cholesterol down to levels not seen in nature. Statins have also been shown to reduce the relative risk of heart attacks in men in their 50's by as much as 30%, but, because heart attacks are relatively rare for this segment of the population, the absolute risk reduction is only on the order of 2%, a point that is often missed by the person being treated.
All drugs have potential side effects, and with any drug it's a matter of weighing the risk/benefit factors to decide whether the drug is warranted. Statin drugs have a remarkably diverse set of side effects, including cognitive and memory impairment, reduced libido, and muscle pain and weakness. The drug manufacturers claim that the incidence of side effects is relatively rare, but often side effects don't appear until after several months or even years into treatment. In many of these cases, it may not be obvious that the statin drug is the cause of the problem. This is especially true because these side effects can easily be attributed to increasing age. In fact, as I will show later, statin side effects can best be interpreted as an acceleration of the aging process.
In my view, statin drugs are never worth the risk of their side effects. Cholesterol is a vital nutrient, without which mammalian cells can not survive, and it is inconceivable to me that crippling the body's ability to synthesize cholesterol can ever be a good idea. In an excellent and highly informative review article published in 2009, Wainwright et al. [43] developed a strong argument that statin drugs, by depleting cholesterol, lead to a destabilization of cell membranes "from head to toe." This problem, in turn, increases risk to a long list of serious health conditions and diseases, including diabetes, multiple sclerosis, cognitive problems, hemorrhagic stroke, cancer, and even ALS (amyotrophic lateral sclerosis, also known commonly as Lou Gehrig's disease). Their arguments are backed up by references to 85 peer-reviewed journal publications. I have argued in previous essays that statins may increase the risk to Alzheimer's disease, as well as to sepsis, cancer, and heart failure.
The most commonly reported side effects to statin therapy are muscle pain and weakness. If left unchecked, these symptoms can progress to rhabdomyolysis (severe muscle damage) and kidney failure. Muscle weakness in the lungs can lead to breathing difficulties; in the heart it leads to heart failure. Statin users are reassured by their doctors that they can halt statin therapy if their liver and muscle enzymes rise too high. In practice, however, it's possible to suffer irreversible muscle damage (the problem does not go away after the statin therapy is stopped), and this can happen even when the enzyme levels are not above the normal range.
This essay will develop an argument for why, over time, a statin user may become increasingly weak, in some cases to the point of major disability. A key message is that muscles are forced to cannibalize themselves to acquire sufficient energy. But another factor is oxidative damage to muscle tissue, with subsequent disintegration of the cell walls. This is true not only for the skeletal muscles, but also for the respiratory muscles controlling breathing and the heart muscle. With continued abuse, the muscle cells disintegrate, and the debris travels in the blood stream to the kidneys, which can lead to kidney failure.
The rest of this essay will unfold as follows. In the next section, I will explain how statin drugs work, which will also show why they interfere with the synthesis not only of cholesterol but also of other essential biological substances involved in cell metabolism. The following section will present evidence that statins damage muscle cells. Sections 4 and 5 describe the biochemical pathways involved in assuring that muscles have enough energy to effect movement, especially during situations of stress such as extreme exercise. Section 6 describes the condition known as rhabdomyolysis, caused by extreme exercise but also by statin drugs, and the subsequent risk to kidney failure. Section 7 describes the role that myoglobin, a key protein found in muscle cells, plays in the disease process. After a section explaining how cholesterol protects cell membranes from oxidative damage, the four subsequent sections (sections 9-12) will be devoted to the repercussions of statin damage to the muscles, the heart, the lungs, and the pancreas, respectively. Finally, the conclusion section will summarize the essay and provide hints about my upcoming essay on ALS, a physically disabling neurodegenerative disease that is due not to muscle damage per se but to damage of the motor neurons in the spinal cord that transmit signals from the brain to the skeletal muscles.
Thursday, February 25, 2010
2. The Biological Mechanism of Statin Drugs
Why do statins cause so many side effects? To answer this question requires explaining all the crucial roles that cholesterol plays in maintaining the integrity and functioning of the body's cells. However, statins interfere not only with the synthesis of cholesterol, but also with the synthesis of an enzyme, coenzyme Q10, that plays a critical role in energy metabolism in all cells. A deficiency in both cholesterol and coenzyme Q10, over time, leads to a huge list of potential health problems. Exactly how an individual responds depends upon their genetic make-up: faced with a deficiency, the body will decide to sacrifice certain cell types in order to safeguard certain other cell types. So, one person may develop Alzheimer's because the brain's neurons are sacrificed, while another succumbs to heart failure or rhabdomyolysis (skeletal muscle wasting).
Statins suppress a critical early step in the multi-step biological pathway that leads to cholesterol synthesis. This is why statins are able to dramatically reduce the blood serum levels of cholesterol. Specifically, statins interfere with the production of the enzyme, HMG-Coenzyme A Reductase, which catalyzes production of mevalonate from its precursor, HMG-Coenzyme A.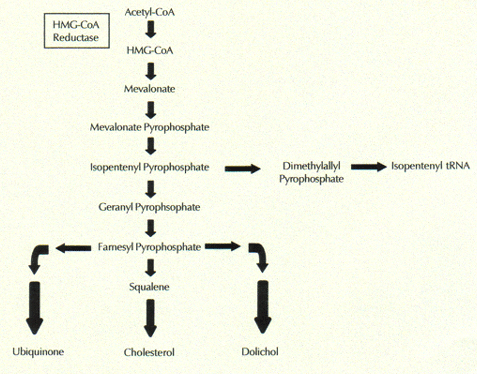 Several more steps produce cholesterol from mevalonate. Mevalonate is also the precursor to a large number of other biologically active molecules that are important for proper cell function. These include the antioxidants, coenzyme Q10 and the dolichols, as shown in the figure to the right.
Several more steps produce cholesterol from mevalonate. Mevalonate is also the precursor to a large number of other biologically active molecules that are important for proper cell function. These include the antioxidants, coenzyme Q10 and the dolichols, as shown in the figure to the right.
The so-called "bad" cholesterol, LDL, delivers cholesterol, fats, and antioxidants from the liver to all the cells of the body. All cells need both fats and cholesterol to maintain healthy membranes, not only in the outer cell wall, but also in the membranes encasing the nucleus, the mitochondria (energy producing units), and the lysosomes (the cell's digestive system). Antioxidants are critical for neutralizing the damaging effects of oxygen exposure, always an issue whenever energy is generated in the mitochondria through a chemical reaction between food sources and oxygen.
In a double-blind placebo controlled study [18], it has been shown that statins can reduce serum levels of coenzyme Q10 by as much as 40%. Coenzyme Q10 is not only a powerful antioxidant, but it also plays a crucial role in the process that breaks down glucose in the presence of oxygen to yield carbon dioxide and water. This metabolic pathway, which is essentially the burning of glucose as fuel, takes place in the mitochondria via the well-known citric acid, or Kreb's cycle. The energy that is released through this process is packaged up in the form of ATP (Adenosine Triphosphate), the currency that all cells use to store their energy reserves.
The dolichols play a special role for the lysosomes [20]. The lysosomes are walled off "rooms" that contain digestive enzymes to break down debris from damaged cell parts so that they can be recycled into useful materials. Lysosomes must maintain a highly acidic internal environment in order for the digestive enzymes to work properly. The dolichols are responsible for pumping hydrogen ions into the lysosomes to keep them highly acidic.
A final way that statins can damage cells is through their entry mechanism. Statins belong to a class of drugs called "amphiphilic" drugs [2] that manage to break through the cell wall in spite of being relatively large. They act like a soap by essentially dissolving a section of the cell membrane. This leaves behind a hole in the wall, which needs to be patched up, as well as debris which must be cleaned up and recycled by the lysosomes. To patch the hole requires new sources of both fats and cholesterol, which come from the LDL particles, whose supply is greatly reduced due to the statin drug. So it becomes increasingly difficult over time for the cell to repair all the holes introduced by the statin drug molecules. As the cell wall becomes more permeable due to previous exposure to an amphiphilic drug, the amount of drug that successfully enters the cell steadily increases over time, leading to ever greater internal concentrations of the drug.
Statins suppress a critical early step in the multi-step biological pathway that leads to cholesterol synthesis. This is why statins are able to dramatically reduce the blood serum levels of cholesterol. Specifically, statins interfere with the production of the enzyme, HMG-Coenzyme A Reductase, which catalyzes production of mevalonate from its precursor, HMG-Coenzyme A.
Several more steps produce cholesterol from mevalonate. Mevalonate is also the precursor to a large number of other biologically active molecules that are important for proper cell function. These include the antioxidants, coenzyme Q10 and the dolichols, as shown in the figure to the right. The so-called "bad" cholesterol, LDL, delivers cholesterol, fats, and antioxidants from the liver to all the cells of the body. All cells need both fats and cholesterol to maintain healthy membranes, not only in the outer cell wall, but also in the membranes encasing the nucleus, the mitochondria (energy producing units), and the lysosomes (the cell's digestive system). Antioxidants are critical for neutralizing the damaging effects of oxygen exposure, always an issue whenever energy is generated in the mitochondria through a chemical reaction between food sources and oxygen.
In a double-blind placebo controlled study [18], it has been shown that statins can reduce serum levels of coenzyme Q10 by as much as 40%. Coenzyme Q10 is not only a powerful antioxidant, but it also plays a crucial role in the process that breaks down glucose in the presence of oxygen to yield carbon dioxide and water. This metabolic pathway, which is essentially the burning of glucose as fuel, takes place in the mitochondria via the well-known citric acid, or Kreb's cycle. The energy that is released through this process is packaged up in the form of ATP (Adenosine Triphosphate), the currency that all cells use to store their energy reserves.
The dolichols play a special role for the lysosomes [20]. The lysosomes are walled off "rooms" that contain digestive enzymes to break down debris from damaged cell parts so that they can be recycled into useful materials. Lysosomes must maintain a highly acidic internal environment in order for the digestive enzymes to work properly. The dolichols are responsible for pumping hydrogen ions into the lysosomes to keep them highly acidic.
A final way that statins can damage cells is through their entry mechanism. Statins belong to a class of drugs called "amphiphilic" drugs [2] that manage to break through the cell wall in spite of being relatively large. They act like a soap by essentially dissolving a section of the cell membrane. This leaves behind a hole in the wall, which needs to be patched up, as well as debris which must be cleaned up and recycled by the lysosomes. To patch the hole requires new sources of both fats and cholesterol, which come from the LDL particles, whose supply is greatly reduced due to the statin drug. So it becomes increasingly difficult over time for the cell to repair all the holes introduced by the statin drug molecules. As the cell wall becomes more permeable due to previous exposure to an amphiphilic drug, the amount of drug that successfully enters the cell steadily increases over time, leading to ever greater internal concentrations of the drug.
3. Statins, Muscle Pain and Weakness, and Rhabdomyolysis
Muscle cells have tremendous energy needs, particularly if the person has been put on an exercise regimen as part of their treatment program. The heart, in particular, never rests. It has to keep on beating 24x7 at the rate of at least once every second. Hence the heart is especially dependent on coenzyme Q10 to replenish the ATP consumed every time it contracts and pushes blood from one chamber to another and out the aorta.
The pharmaceutical industry readily admits that statin therapy may cause muscle pain and/or muscle weakness in some cases, but they claim that the incidence of these side effects is small, on the order of 2%. However, observational studies have shown that at least 10% to 15% of statin users complain of muscle pain [6][40]. The actual number who experience pain or weakness is likely to be much larger, however, because many people are unaware that this is a potential side effect. Furthermore, it sometimes takes several years of cumulative statin damage before the symptoms become intolerable. People are often willing to believe that their aches and pains and generally weakening condition are simply due to getting older.
The reaction by the general community to a relatively benign article posted by WebMD on muscle pain suggests that the problem is much worse than is generally acknowledged. Over 200 often lengthy comments describe many very sad stories; often the doctor was also misinformed, and denied that the pain could be due to the statin drug. A case in point is described in this New York Times article. A woman in Kansas had been taking a statin for years to reduce her cholesterol. Over that same time period, she experienced chronic muscle pain which neither she nor her doctor attributed to the statin therapy. It even led to a useless shoulder operation. Her problem eventually escalated into skin lesions caused by a reaction to toxic protein by-products released by her disintegrating muscles. She was given an antifungal to treat the skin lesions, another misdiagnosis. But the antifungal interacted with the statins [25] to further increase the severity of her muscle disorders. Three months later, she could barely stand, and her pulmonary muscles were so weak she couldn't breathe. She died shortly thereafter.
Rhabdomyolysis is a condition where the muscles rapidly disintegrate due to an injury, often, for example, physical trauma following an accident. But Rhabdomyolysis is also a rare side effect of statins -- essentially where the muscle pain and weakness are extreme. Some people react immediately to statin therapy with severe rhabdomyolysis, and it is often fatal, due to acute renal failure (ARF). Myoglobin is sloughed off from the muscle cells in large amounts, and it overloads the kidneys and causes them to shut down completely. Initiating statin therapy is therefore a bit like Russian roulette -- there is even a known case where a single statin dose caused rhabdomyolysis [21]. One of the statins, Baycol, was abruptly taken off the market in 2001, after 31 people died from subsequent rhabdomyolysis.
The pharmaceutical industry readily admits that statin therapy may cause muscle pain and/or muscle weakness in some cases, but they claim that the incidence of these side effects is small, on the order of 2%. However, observational studies have shown that at least 10% to 15% of statin users complain of muscle pain [6][40]. The actual number who experience pain or weakness is likely to be much larger, however, because many people are unaware that this is a potential side effect. Furthermore, it sometimes takes several years of cumulative statin damage before the symptoms become intolerable. People are often willing to believe that their aches and pains and generally weakening condition are simply due to getting older.
The reaction by the general community to a relatively benign article posted by WebMD on muscle pain suggests that the problem is much worse than is generally acknowledged. Over 200 often lengthy comments describe many very sad stories; often the doctor was also misinformed, and denied that the pain could be due to the statin drug. A case in point is described in this New York Times article. A woman in Kansas had been taking a statin for years to reduce her cholesterol. Over that same time period, she experienced chronic muscle pain which neither she nor her doctor attributed to the statin therapy. It even led to a useless shoulder operation. Her problem eventually escalated into skin lesions caused by a reaction to toxic protein by-products released by her disintegrating muscles. She was given an antifungal to treat the skin lesions, another misdiagnosis. But the antifungal interacted with the statins [25] to further increase the severity of her muscle disorders. Three months later, she could barely stand, and her pulmonary muscles were so weak she couldn't breathe. She died shortly thereafter.
Rhabdomyolysis is a condition where the muscles rapidly disintegrate due to an injury, often, for example, physical trauma following an accident. But Rhabdomyolysis is also a rare side effect of statins -- essentially where the muscle pain and weakness are extreme. Some people react immediately to statin therapy with severe rhabdomyolysis, and it is often fatal, due to acute renal failure (ARF). Myoglobin is sloughed off from the muscle cells in large amounts, and it overloads the kidneys and causes them to shut down completely. Initiating statin therapy is therefore a bit like Russian roulette -- there is even a known case where a single statin dose caused rhabdomyolysis [21]. One of the statins, Baycol, was abruptly taken off the market in 2001, after 31 people died from subsequent rhabdomyolysis.
4. How Muscles Maintain their Energy Supply
In this section and the next, I will describe the metabolic pathways involved in assuring that muscle cells have enough energy to contract. When oxygen is available, and when it can be safely exploited, the muscle can decompose food sources into carbon dioxide and water, by consuming the oxygen. But oxygen, while life-giving, is also a very dangerous substance, and if the process is not orchestrated exactly according to plan, there can be a lot of collateral damage due to the wrong substances reacting with the oxygen. As you will see later, myoglobin, which is responsible for buffering oxygen and delivering it from the cell wall to the mitochondria, picks up a lot of the collateral damage. This aerobic metabolic process is termed respiration, and it takes place inside special organelles called mitochondria, whose sole responsibility is to digest foods and generate energy for the cell.
Whenever oxygen is in short supply, or if the mitochondria are dysfunctional, the cell has alternative ways to generate energy (e.g., fermentation), which take place, in the absence of oxygen, in the main compartment of the cell, called the cytoplasm. These processes require exchanges of nutrients between the muscles and the liver, and they require the assistance of special enzymes which then show up in the blood stream. These are the very same enzymes whose concentrations are monitored to detect whether a statin drug may have damaged the muscles.
If you don't feel compelled to know the details of how all these processes work, you could skip this section and section 5, and, I think, still be able to follow the rest of the story.
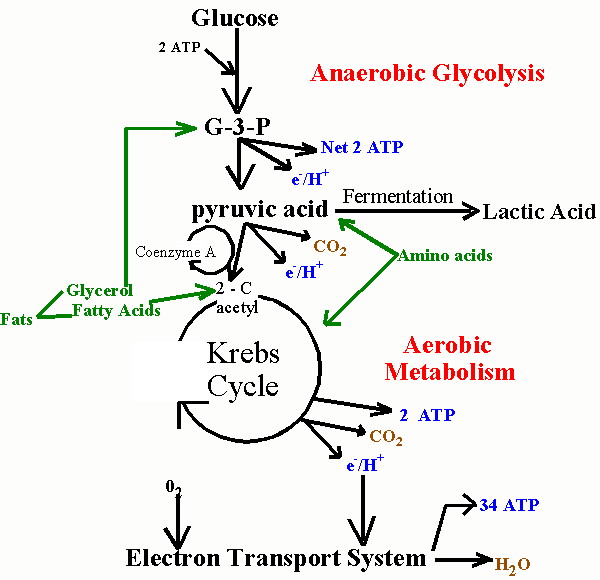 In order to explain how statins damage muscles, I will first need to explain how muscles manage their energy needs. Muscles require a significant amount of energy to contract, and they get most of this energy by breaking down fatty acids and glucose obtained originally from food sources. Like all eukaryotic cells (cells containing a nucleus), muscle cells are able to generate a lot of energy through aerobic (oxygen-requiring) processes that are sequestered within special energy-generating subregions of the cell called mitochondria. This aerobic metabolic process is highly efficient, generating as many as 30 units of ATP (Adenosine Triphosphate) for each molecule of glucose. ATP can be thought of as an energy currency, because it can be easily broken down to AMP (adenosine monophosphate), releasing the stored energy in the process, which will then fuel cell contraction.
In order to explain how statins damage muscles, I will first need to explain how muscles manage their energy needs. Muscles require a significant amount of energy to contract, and they get most of this energy by breaking down fatty acids and glucose obtained originally from food sources. Like all eukaryotic cells (cells containing a nucleus), muscle cells are able to generate a lot of energy through aerobic (oxygen-requiring) processes that are sequestered within special energy-generating subregions of the cell called mitochondria. This aerobic metabolic process is highly efficient, generating as many as 30 units of ATP (Adenosine Triphosphate) for each molecule of glucose. ATP can be thought of as an energy currency, because it can be easily broken down to AMP (adenosine monophosphate), releasing the stored energy in the process, which will then fuel cell contraction.
Unfortunately, the process of metabolizing food sources for energy is quite complex. I have found two images which depict food metabolism in complementary ways, where one (above, right) shows chemical reactions and the other (below, left) schematizes the regions of the cell that are involved. They use slightly different nomenclature, but I will try to link them together when necessary. When glucose first enters the cell (mediated through insulin), it is converted to pyruvate (also called pyruvic acid) in the cell's cytoplasm (the main compartment of the cell). This process releases a small amount of ATP, but does not require oxygen, which makes it useful when oxygen is in short supply. The pyruvate can also be broken down to lactate (also called lactic acid) (fermentation, oxygen absent in the figure below) in the cytoplasm, without requiring any oxygen, so-called anaerobic metabolism, to release additional energy. This pathway is important to muscle cells under conditions of extreme exercise, when oxygen supplies become depleted.
To generate a much larger amount of ATP requires the help of the mitochondria (the large oval purple-shaped object in the figure), and involves a well known process referred to multiple ways: as the respiratory or electron transport chain, the Tricarboxylic acid (TCA) cycle or the Krebs Cycle (TCA cycle, oxygen present in the figure below; Krebs Cycle, Aerobic Metabolism in the figure above). The process is tricky, because oxygen molecules (O2) have to be split apart, and, during intermediate stages, dangerous free radicals are lying around (individual negatively charged oxygen atoms that have not yet fully combined with hydrogen (H+) to form the very stable molecule, water (H2O)).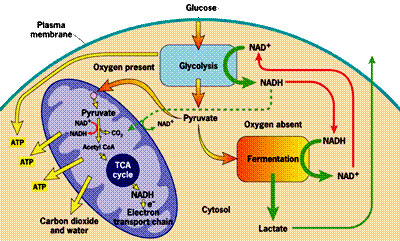 These free radicals are highly reactive. Antioxidants are compounds that can absorb these free radicals and render them harmless. Two very important antioxidants that play a critical role in the electron transport chain are coenzyme Q10 (also known as ubiquinone) and cytochrome c.
These free radicals are highly reactive. Antioxidants are compounds that can absorb these free radicals and render them harmless. Two very important antioxidants that play a critical role in the electron transport chain are coenzyme Q10 (also known as ubiquinone) and cytochrome c.
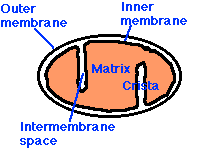 The small figure to the left shows a schematic of a mitochondrion, and the larger figure below shows a more detailed explanation of the electron transport chain process that takes place along the wall enclosing the mitochondrion, generating a large percentage of the cell's energy needs in the process. The electron transport chain injects protons (H+) into the intermembrane space, essentially creating a battery (charge differential across the membrane) that can then complete the process of converting (spent) AMP back to ATP as a renewed energy source. If there is an insufficient supply of coenzyme Q10 (also known as "ubiquinone", the "Q" in the figure), then the
The small figure to the left shows a schematic of a mitochondrion, and the larger figure below shows a more detailed explanation of the electron transport chain process that takes place along the wall enclosing the mitochondrion, generating a large percentage of the cell's energy needs in the process. The electron transport chain injects protons (H+) into the intermembrane space, essentially creating a battery (charge differential across the membrane) that can then complete the process of converting (spent) AMP back to ATP as a renewed energy source. If there is an insufficient supply of coenzyme Q10 (also known as "ubiquinone", the "Q" in the figure), then the 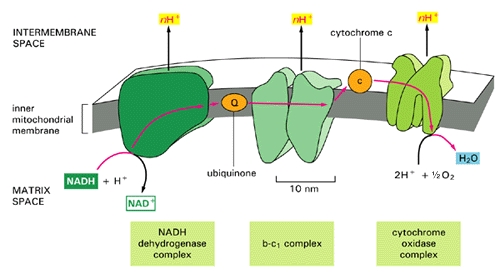 electron transport chain will not work as efficiently. Hydrogen ions will leak back into the mitochondrion through a passive process, requiring a much larger expenditure of energy to push them back out [20]. The battery charge will be reduced, and there will be a decrease in the amount of ATP that can be generated. The net effect will be very similar to the effect of insufficient oxygen, with respect to energy generated. However, it will be much more damaging because, instead of being absent, the oxygen is present but is only partially converted to water (2H+ + 1/2 O2 -> H2O at the right of the figure), since the chain of events is held up at the "Q" position. Various highly toxic charged ions containing oxygen, such as -OH, H2O2 (hydrogen peroxide) and *OH, will linger and wreak havoc on the muscle cell, as you will see later.
electron transport chain will not work as efficiently. Hydrogen ions will leak back into the mitochondrion through a passive process, requiring a much larger expenditure of energy to push them back out [20]. The battery charge will be reduced, and there will be a decrease in the amount of ATP that can be generated. The net effect will be very similar to the effect of insufficient oxygen, with respect to energy generated. However, it will be much more damaging because, instead of being absent, the oxygen is present but is only partially converted to water (2H+ + 1/2 O2 -> H2O at the right of the figure), since the chain of events is held up at the "Q" position. Various highly toxic charged ions containing oxygen, such as -OH, H2O2 (hydrogen peroxide) and *OH, will linger and wreak havoc on the muscle cell, as you will see later.
There are a number of rare genetic disorders that involve mutations in genes encoding enzymes that operate within the electron transport chain [23][32]. Particularly relevant to our story are Complex I enzymes, because Coenzyme Q10 is one of them. An interesting case study involved two sisters [23], both of whom suffered from a genetic mutation leading to a defect identified to be associated with the NADH-Coenzyme Q10 complex. As would be predicted, they suffered from substantially decreased rates of respiratory metabolism (the process discussed above). They also were extremely weak and had marked intolerance to exercise. When they exercised, their levels of lactate and pyruvate rose sharply in the blood, an indication that they were relying on anaerobic fermentation in the cytoplasm rather than aerobic metabolism in the mitochondria to meet their energy needs.
Whenever oxygen is in short supply, or if the mitochondria are dysfunctional, the cell has alternative ways to generate energy (e.g., fermentation), which take place, in the absence of oxygen, in the main compartment of the cell, called the cytoplasm. These processes require exchanges of nutrients between the muscles and the liver, and they require the assistance of special enzymes which then show up in the blood stream. These are the very same enzymes whose concentrations are monitored to detect whether a statin drug may have damaged the muscles.
If you don't feel compelled to know the details of how all these processes work, you could skip this section and section 5, and, I think, still be able to follow the rest of the story.
In order to explain how statins damage muscles, I will first need to explain how muscles manage their energy needs. Muscles require a significant amount of energy to contract, and they get most of this energy by breaking down fatty acids and glucose obtained originally from food sources. Like all eukaryotic cells (cells containing a nucleus), muscle cells are able to generate a lot of energy through aerobic (oxygen-requiring) processes that are sequestered within special energy-generating subregions of the cell called mitochondria. This aerobic metabolic process is highly efficient, generating as many as 30 units of ATP (Adenosine Triphosphate) for each molecule of glucose. ATP can be thought of as an energy currency, because it can be easily broken down to AMP (adenosine monophosphate), releasing the stored energy in the process, which will then fuel cell contraction.Unfortunately, the process of metabolizing food sources for energy is quite complex. I have found two images which depict food metabolism in complementary ways, where one (above, right) shows chemical reactions and the other (below, left) schematizes the regions of the cell that are involved. They use slightly different nomenclature, but I will try to link them together when necessary. When glucose first enters the cell (mediated through insulin), it is converted to pyruvate (also called pyruvic acid) in the cell's cytoplasm (the main compartment of the cell). This process releases a small amount of ATP, but does not require oxygen, which makes it useful when oxygen is in short supply. The pyruvate can also be broken down to lactate (also called lactic acid) (fermentation, oxygen absent in the figure below) in the cytoplasm, without requiring any oxygen, so-called anaerobic metabolism, to release additional energy. This pathway is important to muscle cells under conditions of extreme exercise, when oxygen supplies become depleted.
To generate a much larger amount of ATP requires the help of the mitochondria (the large oval purple-shaped object in the figure), and involves a well known process referred to multiple ways: as the respiratory or electron transport chain, the Tricarboxylic acid (TCA) cycle or the Krebs Cycle (TCA cycle, oxygen present in the figure below; Krebs Cycle, Aerobic Metabolism in the figure above). The process is tricky, because oxygen molecules (O2) have to be split apart, and, during intermediate stages, dangerous free radicals are lying around (individual negatively charged oxygen atoms that have not yet fully combined with hydrogen (H+) to form the very stable molecule, water (H2O)).
These free radicals are highly reactive. Antioxidants are compounds that can absorb these free radicals and render them harmless. Two very important antioxidants that play a critical role in the electron transport chain are coenzyme Q10 (also known as ubiquinone) and cytochrome c. The small figure to the left shows a schematic of a mitochondrion, and the larger figure below shows a more detailed explanation of the electron transport chain process that takes place along the wall enclosing the mitochondrion, generating a large percentage of the cell's energy needs in the process. The electron transport chain injects protons (H+) into the intermembrane space, essentially creating a battery (charge differential across the membrane) that can then complete the process of converting (spent) AMP back to ATP as a renewed energy source. If there is an insufficient supply of coenzyme Q10 (also known as "ubiquinone", the "Q" in the figure), then the electron transport chain will not work as efficiently. Hydrogen ions will leak back into the mitochondrion through a passive process, requiring a much larger expenditure of energy to push them back out [20]. The battery charge will be reduced, and there will be a decrease in the amount of ATP that can be generated. The net effect will be very similar to the effect of insufficient oxygen, with respect to energy generated. However, it will be much more damaging because, instead of being absent, the oxygen is present but is only partially converted to water (2H+ + 1/2 O2 -> H2O at the right of the figure), since the chain of events is held up at the "Q" position. Various highly toxic charged ions containing oxygen, such as -OH, H2O2 (hydrogen peroxide) and *OH, will linger and wreak havoc on the muscle cell, as you will see later. There are a number of rare genetic disorders that involve mutations in genes encoding enzymes that operate within the electron transport chain [23][32]. Particularly relevant to our story are Complex I enzymes, because Coenzyme Q10 is one of them. An interesting case study involved two sisters [23], both of whom suffered from a genetic mutation leading to a defect identified to be associated with the NADH-Coenzyme Q10 complex. As would be predicted, they suffered from substantially decreased rates of respiratory metabolism (the process discussed above). They also were extremely weak and had marked intolerance to exercise. When they exercised, their levels of lactate and pyruvate rose sharply in the blood, an indication that they were relying on anaerobic fermentation in the cytoplasm rather than aerobic metabolism in the mitochondria to meet their energy needs.
5. Managing Energy Needs During Extreme Exercise
Whenever a healthy person experiences extreme exercise, such as running a 500 meter dash, the muscles are challenged to come up with sufficient ATP to satisfy their energy needs. Both oxygen and glucose can become depleted. To compensate for these deficiencies, muscle cells have devised a complex set of strategies, which operate in the cytoplasm instead of in the mitochondria. They involve a number of enzymes that will resurface later in our story, since they are the enzymes that are monitored to determine whether statin drugs are damaging muscles and/or the liver.
As you've already seen, one option is to generate lactic acid anaerobically (without consuming oxygen), but this still provides only 1/6 as much ATP as the aerobic process taking place in the mitochondria. The process of generating energy from ATP happens in two steps: ATP is first converted to ADP (adenosine diphosphate) and finally to AMP (adenosine monophosphate). When excess amounts of AMP accumulate in the muscle cell, the cell is induced to take up extra glucose, which will soon deplete the supply (of glucose) in the blood unless the liver can efficiently regenerate more. ADP can be converted back to ATP with the help of an enzyme, creatine kinase. In addition, the conversion of pyruvate (generated anaerobically from glucose) to lactate requires the help of another enzyme, lactate dehydrogenase. Lactate builds up when oxygen is insufficient, and is released into the blood stream. Fortunately, the heart is able to utilize lactate as an alternative fuel source [9], which becomes especially important during times of extreme exercise.
In order for the liver to generate more glucose, it needs a substrate. In the short term, muscles can supply this substrate, but it requires self cannibalization. During brief periods of starvation, human muscle cells adapt quickly by breaking down muscle proteins and converting them into a basic amino acid, alanine [34]. Muscles then rely on a novel mechanism that involves an exchange system with the liver, the so-called glucose-alanine cycle. The alanine, derived from muscle protein, is released into the blood and shipped to the liver to be utilized for energy generation, as shown in the accompanying figure. The liver can then generate more glucose from the alanine through gluconeogenesis, while exporting the waste product, urea, to the kidneys for excretion. This also allows the liver to regenerate some ATP to help satisfy its own energy needs, which are very large during such stressful conditions. The glucose is shipped through the blood stream to the muscle cell, which eagerly takes it up to generate more ATP for itself. The anaerobic processing of glucose yields pyruvate which can also be turned into alanine, but it needs yet another enzyme to work. Thus, whenever pyruvate can't be sent to the mitochondria because of insufficient oxygen, it can instead be converted to alanine with the help of an enzyme, ALT (alanine aminotransferase),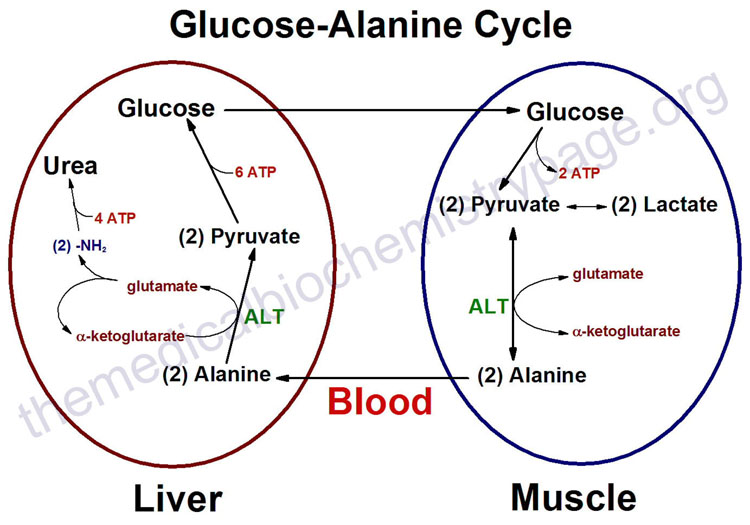 as long as there is a good supply of glutamate, which is converted to alpha-keto glutarate in the process.
as long as there is a good supply of glutamate, which is converted to alpha-keto glutarate in the process.
In the above discussion, several enzymes have been identified that must be present for these cytoplasmic energy-generating processes to function. These include creatine kinase, lactate dehydrogenase, and ALT. The so-called liver enzyme test that is conducted routinely with statin users measures the concentration of ALT in the blood. Muscle enzyme tests detect creatine kinase and lactate dehydrogenase concentrations in the blood. So these tests are all measuring these particular enzymes because they signal that muscles are preferentially processing glucose anaerobically in the cytoplasm instead of aerobically in the mitochondria; i.e., the mitochondria are not working properly. You should keep this point in mind as we will revisit it later on.
As you've already seen, one option is to generate lactic acid anaerobically (without consuming oxygen), but this still provides only 1/6 as much ATP as the aerobic process taking place in the mitochondria. The process of generating energy from ATP happens in two steps: ATP is first converted to ADP (adenosine diphosphate) and finally to AMP (adenosine monophosphate). When excess amounts of AMP accumulate in the muscle cell, the cell is induced to take up extra glucose, which will soon deplete the supply (of glucose) in the blood unless the liver can efficiently regenerate more. ADP can be converted back to ATP with the help of an enzyme, creatine kinase. In addition, the conversion of pyruvate (generated anaerobically from glucose) to lactate requires the help of another enzyme, lactate dehydrogenase. Lactate builds up when oxygen is insufficient, and is released into the blood stream. Fortunately, the heart is able to utilize lactate as an alternative fuel source [9], which becomes especially important during times of extreme exercise.
In order for the liver to generate more glucose, it needs a substrate. In the short term, muscles can supply this substrate, but it requires self cannibalization. During brief periods of starvation, human muscle cells adapt quickly by breaking down muscle proteins and converting them into a basic amino acid, alanine [34]. Muscles then rely on a novel mechanism that involves an exchange system with the liver, the so-called glucose-alanine cycle. The alanine, derived from muscle protein, is released into the blood and shipped to the liver to be utilized for energy generation, as shown in the accompanying figure. The liver can then generate more glucose from the alanine through gluconeogenesis, while exporting the waste product, urea, to the kidneys for excretion. This also allows the liver to regenerate some ATP to help satisfy its own energy needs, which are very large during such stressful conditions. The glucose is shipped through the blood stream to the muscle cell, which eagerly takes it up to generate more ATP for itself. The anaerobic processing of glucose yields pyruvate which can also be turned into alanine, but it needs yet another enzyme to work. Thus, whenever pyruvate can't be sent to the mitochondria because of insufficient oxygen, it can instead be converted to alanine with the help of an enzyme, ALT (alanine aminotransferase),
as long as there is a good supply of glutamate, which is converted to alpha-keto glutarate in the process. In the above discussion, several enzymes have been identified that must be present for these cytoplasmic energy-generating processes to function. These include creatine kinase, lactate dehydrogenase, and ALT. The so-called liver enzyme test that is conducted routinely with statin users measures the concentration of ALT in the blood. Muscle enzyme tests detect creatine kinase and lactate dehydrogenase concentrations in the blood. So these tests are all measuring these particular enzymes because they signal that muscles are preferentially processing glucose anaerobically in the cytoplasm instead of aerobically in the mitochondria; i.e., the mitochondria are not working properly. You should keep this point in mind as we will revisit it later on.
6. Extreme Exercise can lead to Rhabdomyolysis
When people engage in extreme exercise such as long distance marathon running or weight-bearing exercises, they run the risk of causing serious damage, both to their muscles and to their kidneys, due to the stress imposed on their system in trying to maintain adequate energy to fuel the muscles. It has become common practice to measure the levels of creatine kinase in the blood as a known indicator of potential damage [5]. A person whose creatine kinase level gets alarmingly high will likely need immediate medical attention to avoid renal [kidney] failure.
The cause of the kidney failure is most likely myoglobin that has been dumped into the blood stream by compromised or dead muscle cells, due to rhabdomyolysis. If too much myoglobin is released, particularly with inadequate water supply, the myoglobin can block the renal filtration system causing a condition known as "acute tubular necrosis." The problem can be easily detected by observing the color of the urine, which will be dark brown. A study published in 2009 showed that, in rhabdomyolysis, the damage to the kidneys involves direct interaction between myoglobin and the mitochondria in the kidney cells [33]. The resulting oxidation of the mitochondrial membranes leads to respiratory failure and subsequent cell death.
Myoglobinuria is the term used to describe the presence of myoglobin in the urine, usually due to rhabdomyolysis. According to [37], 15% of patients with severe myoglobinuria develop acute renal failure, and it is associated with high mortality rates. Dialysis or intravenous fluids must be introduced fast enough, or the person will not be able to recover.
As early as 1991, a group of Japanese researchers [38] demonstrated that coenzyme Q10 could be administered orally to protect rats from muscle damage due to strenuous exercise. They also noticed that rats that were administered coenzyme Q10 did not have elevated levels of creatine kinase and lactate dehydrogenase, whereas the control rats did.
The cause of the kidney failure is most likely myoglobin that has been dumped into the blood stream by compromised or dead muscle cells, due to rhabdomyolysis. If too much myoglobin is released, particularly with inadequate water supply, the myoglobin can block the renal filtration system causing a condition known as "acute tubular necrosis." The problem can be easily detected by observing the color of the urine, which will be dark brown. A study published in 2009 showed that, in rhabdomyolysis, the damage to the kidneys involves direct interaction between myoglobin and the mitochondria in the kidney cells [33]. The resulting oxidation of the mitochondrial membranes leads to respiratory failure and subsequent cell death.
Myoglobinuria is the term used to describe the presence of myoglobin in the urine, usually due to rhabdomyolysis. According to [37], 15% of patients with severe myoglobinuria develop acute renal failure, and it is associated with high mortality rates. Dialysis or intravenous fluids must be introduced fast enough, or the person will not be able to recover.
As early as 1991, a group of Japanese researchers [38] demonstrated that coenzyme Q10 could be administered orally to protect rats from muscle damage due to strenuous exercise. They also noticed that rats that were administered coenzyme Q10 did not have elevated levels of creatine kinase and lactate dehydrogenase, whereas the control rats did.
7. Myoglobin: The Good, The Bad, and the Ugly
Myoglobin is a unique protein specially adapted for muscle cells to help them with their enormous needs for oxygen. Its physical structure is schematized in the figure on the right. It resembles hemoglobin in that it contains a central heme element (schematized in red in the figure) whose active 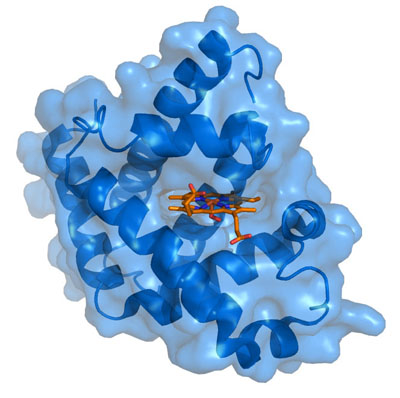 ingredient is a single charged iron atom (Fe). While hemoglobin, found in red blood cells, transports oxygen from the lungs to all the tissues of the body, myoglobin functions to store excess oxygen within the muscle cell, to help buffer supply during periods of excessive demand. It also transports oxygen from the cell wall to the mitochondria. Even with help from the myoglobin, it's often the case that muscles have to resort to anaerobic metabolism under strenuous exercise, during which lactic acid is built up and released into the blood stream.
ingredient is a single charged iron atom (Fe). While hemoglobin, found in red blood cells, transports oxygen from the lungs to all the tissues of the body, myoglobin functions to store excess oxygen within the muscle cell, to help buffer supply during periods of excessive demand. It also transports oxygen from the cell wall to the mitochondria. Even with help from the myoglobin, it's often the case that muscles have to resort to anaerobic metabolism under strenuous exercise, during which lactic acid is built up and released into the blood stream.
Myoglobin exists in at least three distinct forms, which can be characterized as Mg+2 (Ferrous), Mg+3 (Ferric), and Mg+4 (Ferryl), depending upon the amount of charge that is present on the central Iron atom. As Mg+2, its healthy state, it will readily take up oxygen and store it, whereas, when converted to Mg+3 by the addition of a proton, it becomes inert. However, with the addition of yet another proton, it becomes Mg+4, a highly toxic reactive agent that will begin to break down the fatty acids contained in the outer cell wall of the muscle cell (so-called peroxidative damage)[35], and go on to destroy the cholesterol in the cell wall as well [31]. Myoglobin becomes Ferryl myoglobin in the presence of excess amounts of free radicals, i.e., under oxidative stress induced by highly reactive oxygen compounds like hydrogen peroxide. Recall that, with statin therapy, hydrogen peroxide is generated in the mitochondria because the process of breaking down oxygen and converting it to water is incomplete -- due to the insufficient supply of coenzyme Q10.
An excellent article describing the process by which a cell is injured by oxidative stress was written by John Farber in 1994 [13]. He wrote: "All aerobic cells generate, enzymatically or nonenzymatically, a constitutive flux of O2-, H2O2, and possibly *OH. At the same time, the abundant antioxidant defenses of most cells, again both enzymatic and nonenzymatic, prevent these species from causing cell injury. Nevertheless, there are situations in which the rate of formation of partially reduced oxygen species is increased and/or the antioxidant defenses of the cells are weakened. In either case, oxidative cell injury may result." [14, p. 17]. The process of aerobic oxidation of food sources to generate energy is confined to the mitochondria in order to protect the constituents in the cytoplasm as much as possible. But myoglobin is tasked with transporting oxygen from the cell wall through the cytoplasm to the mitochondria. It can't avoid oxygen exposure, and when it delivers the oxygen, it necessarily has to come in contact with these toxic intermediate products of the process that ultimately converts oxygen to water. One of the most important roles of coenzyme Q10 in the muscle cells is to neutralize the damage to myoglobin caused by these oxidative agents.
When a person suffers from a heart attack (ischemic event), their muscles experience an extreme lack of oxygen due to the heart's temporary inability to pump blood. However, one of the most dangerous aspects of a heart attack is the so-called reperfusion period, when blood circulation is restored, but after the cells have suffered injury as a consequence of oxygen deprivation [29]. This condition is especially problematic for the heart muscle, since it is so crucial to survival. In a study involving rats that had suffered from heart attacks, it was proposed that the injury is a direct consequence of exposure to the Fe+4 form of myoglobin (ferryl myoglobin) [1]. Because the cells have been unable to maintain their physiological state during the deprivation period, they are highly vulnerable to oxidative stress.
Once the fatty acids in a muscle's cell wall are broken down due to exposure to toxic Ferryl myoglobin, the cell rapidly disintegrates. Because the cell wall is no longer impermeable to ions, large amounts of calcium start rushing into the cell, and soon after it dies [14]. The debris of the dead and dying cells gets dispersed into the blood stream, and makes its way to the kidneys for disposal. This causes a tremendous load on the kidneys which can sometimes lead to their failure as well [47], and the situation cascades into a downward spiral.
In 1994, Mordente et al. published a paper that investigated in vitro the degree to which coenzyme Q could protect myoglobin from oxidative damage [28]. Their results showed convincingly that coenzyme Q can work as a natural antioxidant for myoglobin. To quote the last sentence of their abstract: "Collectively, these studies suggest that the proposed function of coenzyme Q as a naturally occurring antioxidant might well relate to its ability of reducing H2O2 [hydrogen peroxide]-activated myoglobin. Coenzyme Q should therefore mitigate cardiac or muscular dysfunctions that are caused by an abnormal generation of H2O2."
ingredient is a single charged iron atom (Fe). While hemoglobin, found in red blood cells, transports oxygen from the lungs to all the tissues of the body, myoglobin functions to store excess oxygen within the muscle cell, to help buffer supply during periods of excessive demand. It also transports oxygen from the cell wall to the mitochondria. Even with help from the myoglobin, it's often the case that muscles have to resort to anaerobic metabolism under strenuous exercise, during which lactic acid is built up and released into the blood stream. Myoglobin exists in at least three distinct forms, which can be characterized as Mg+2 (Ferrous), Mg+3 (Ferric), and Mg+4 (Ferryl), depending upon the amount of charge that is present on the central Iron atom. As Mg+2, its healthy state, it will readily take up oxygen and store it, whereas, when converted to Mg+3 by the addition of a proton, it becomes inert. However, with the addition of yet another proton, it becomes Mg+4, a highly toxic reactive agent that will begin to break down the fatty acids contained in the outer cell wall of the muscle cell (so-called peroxidative damage)[35], and go on to destroy the cholesterol in the cell wall as well [31]. Myoglobin becomes Ferryl myoglobin in the presence of excess amounts of free radicals, i.e., under oxidative stress induced by highly reactive oxygen compounds like hydrogen peroxide. Recall that, with statin therapy, hydrogen peroxide is generated in the mitochondria because the process of breaking down oxygen and converting it to water is incomplete -- due to the insufficient supply of coenzyme Q10.
An excellent article describing the process by which a cell is injured by oxidative stress was written by John Farber in 1994 [13]. He wrote: "All aerobic cells generate, enzymatically or nonenzymatically, a constitutive flux of O2-, H2O2, and possibly *OH. At the same time, the abundant antioxidant defenses of most cells, again both enzymatic and nonenzymatic, prevent these species from causing cell injury. Nevertheless, there are situations in which the rate of formation of partially reduced oxygen species is increased and/or the antioxidant defenses of the cells are weakened. In either case, oxidative cell injury may result." [14, p. 17]. The process of aerobic oxidation of food sources to generate energy is confined to the mitochondria in order to protect the constituents in the cytoplasm as much as possible. But myoglobin is tasked with transporting oxygen from the cell wall through the cytoplasm to the mitochondria. It can't avoid oxygen exposure, and when it delivers the oxygen, it necessarily has to come in contact with these toxic intermediate products of the process that ultimately converts oxygen to water. One of the most important roles of coenzyme Q10 in the muscle cells is to neutralize the damage to myoglobin caused by these oxidative agents.
When a person suffers from a heart attack (ischemic event), their muscles experience an extreme lack of oxygen due to the heart's temporary inability to pump blood. However, one of the most dangerous aspects of a heart attack is the so-called reperfusion period, when blood circulation is restored, but after the cells have suffered injury as a consequence of oxygen deprivation [29]. This condition is especially problematic for the heart muscle, since it is so crucial to survival. In a study involving rats that had suffered from heart attacks, it was proposed that the injury is a direct consequence of exposure to the Fe+4 form of myoglobin (ferryl myoglobin) [1]. Because the cells have been unable to maintain their physiological state during the deprivation period, they are highly vulnerable to oxidative stress.
Once the fatty acids in a muscle's cell wall are broken down due to exposure to toxic Ferryl myoglobin, the cell rapidly disintegrates. Because the cell wall is no longer impermeable to ions, large amounts of calcium start rushing into the cell, and soon after it dies [14]. The debris of the dead and dying cells gets dispersed into the blood stream, and makes its way to the kidneys for disposal. This causes a tremendous load on the kidneys which can sometimes lead to their failure as well [47], and the situation cascades into a downward spiral.
In 1994, Mordente et al. published a paper that investigated in vitro the degree to which coenzyme Q could protect myoglobin from oxidative damage [28]. Their results showed convincingly that coenzyme Q can work as a natural antioxidant for myoglobin. To quote the last sentence of their abstract: "Collectively, these studies suggest that the proposed function of coenzyme Q as a naturally occurring antioxidant might well relate to its ability of reducing H2O2 [hydrogen peroxide]-activated myoglobin. Coenzyme Q should therefore mitigate cardiac or muscular dysfunctions that are caused by an abnormal generation of H2O2."
8. How Cholesterol Protects Membranes and Saves Energy
Mammalian cells can not survive without cholesterol [45]. Cholesterol is found in the outer wall (cell membrane) of all cells in the body. It is also found in internal membranes that surround both the mitochondria and the lysosomes (highly acidic containers of digestive 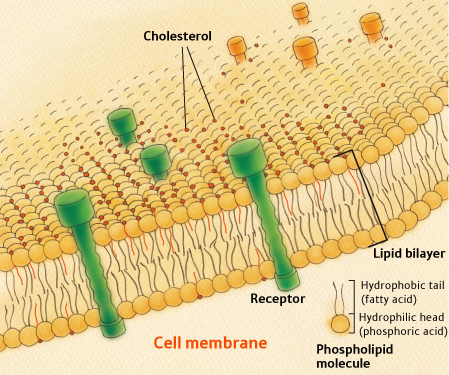 enzymes). To understand how cholesterol works, you need to know something about the structure of cell membranes. All cell membranes are built from a so-called lipid bilayer, as illustrated in the figure to the right. The lipid bilayer contains two parallel chains of phospholipids (the same phospholipids that encase LDL particles, the so-called "bad" cholesterol). Phospholipids have the unique property that one end of the molecule is hydrophobic (water insoluble) and the other is hydrophilic (water soluble). The two chains in the lipid bilayer orient themselves such that the hydrophobic sides of both layers are adjoining in the center of the membrane. This central hydrophobic layer thus contains fatty acids that are vulnerable to oxidative damage. The outer parts, facing both the exterior and the interior of the cell, are water-soluble. Cholesterol molecules are dispersed throughout the membrane at strategic locations.
enzymes). To understand how cholesterol works, you need to know something about the structure of cell membranes. All cell membranes are built from a so-called lipid bilayer, as illustrated in the figure to the right. The lipid bilayer contains two parallel chains of phospholipids (the same phospholipids that encase LDL particles, the so-called "bad" cholesterol). Phospholipids have the unique property that one end of the molecule is hydrophobic (water insoluble) and the other is hydrophilic (water soluble). The two chains in the lipid bilayer orient themselves such that the hydrophobic sides of both layers are adjoining in the center of the membrane. This central hydrophobic layer thus contains fatty acids that are vulnerable to oxidative damage. The outer parts, facing both the exterior and the interior of the cell, are water-soluble. Cholesterol molecules are dispersed throughout the membrane at strategic locations.
An article published in 2009 by Kucerka et al. [22] nicely sums up several known roles of cholesterol in membranes: "Cholesterol is found in all animal cell membranes and is required for proper membrane permeability and fluidity. It is also needed for building and maintaining cell membranes, and may act as an antioxidant. Recently, cholesterol has also been implicated in cell signaling processes, and is suggested to enable lipid raft formation in the plasma membrane." [Ibid, p. 16358] The article goes on to describe how cholesterol is able to orient itself within the membrane either vertically (bridging across the membrane) or horizontally (sequestered within the hydrophobic central space of the membrane lipid bilayer). How it is oriented depends upon the degree to which the fatty acids in the membrane are saturated, with saturated fatty acids greatly favoring the vertical over the horizontal orientation. Cholesterol can also flip easily from one side of the bilayer to the other. All of this flexibility in its orientation within the membrane allows it to operate effectively as a signaling molecule.
A fascinating article written by Thomas Haines in 2001 proposes a novel but compelling role for cholesterol in protecting the cell membrane from sodium leaks [20]. All mammalian cells maintain an ion gradient across their outer wall, which is utilized to fuel cell chemical processes. The so-called sodium pump is an active process that constantly pumps sodium out of the cell to maintain this charge difference. The pump consumes ATP in the process. Working against the pump is a passive leakage mechanism that causes sodium to drift back into the cell. To the extent that the membrane can be constructed to resist leakage (kind of like putting insulation in the attic of a house), it will require less ATP to maintain sodium concentrations appropriate for the cell to function properly.
The Haines article argues that cholesterol plays an essential role in protecting the cell wall from sodium leakage. Sodium leakage is a much bigger problem (it leaks 7 to 11 times as fast in the absence of cholesterol) for unsaturated fatty acids as for saturated fatty acids [4]. However, unsaturated fatty acids also encourage cholesterol to arrange itself in the central layer. By accumulating there, it provides extra insulation preventing the charged sodium ions from passively hopping from the exterior to the interior of the cell. Other experiments [30] have shown that the relative sodium leakage rates are reduced by 300% in the presence of cholesterol.
enzymes). To understand how cholesterol works, you need to know something about the structure of cell membranes. All cell membranes are built from a so-called lipid bilayer, as illustrated in the figure to the right. The lipid bilayer contains two parallel chains of phospholipids (the same phospholipids that encase LDL particles, the so-called "bad" cholesterol). Phospholipids have the unique property that one end of the molecule is hydrophobic (water insoluble) and the other is hydrophilic (water soluble). The two chains in the lipid bilayer orient themselves such that the hydrophobic sides of both layers are adjoining in the center of the membrane. This central hydrophobic layer thus contains fatty acids that are vulnerable to oxidative damage. The outer parts, facing both the exterior and the interior of the cell, are water-soluble. Cholesterol molecules are dispersed throughout the membrane at strategic locations.An article published in 2009 by Kucerka et al. [22] nicely sums up several known roles of cholesterol in membranes: "Cholesterol is found in all animal cell membranes and is required for proper membrane permeability and fluidity. It is also needed for building and maintaining cell membranes, and may act as an antioxidant. Recently, cholesterol has also been implicated in cell signaling processes, and is suggested to enable lipid raft formation in the plasma membrane." [Ibid, p. 16358] The article goes on to describe how cholesterol is able to orient itself within the membrane either vertically (bridging across the membrane) or horizontally (sequestered within the hydrophobic central space of the membrane lipid bilayer). How it is oriented depends upon the degree to which the fatty acids in the membrane are saturated, with saturated fatty acids greatly favoring the vertical over the horizontal orientation. Cholesterol can also flip easily from one side of the bilayer to the other. All of this flexibility in its orientation within the membrane allows it to operate effectively as a signaling molecule.
A fascinating article written by Thomas Haines in 2001 proposes a novel but compelling role for cholesterol in protecting the cell membrane from sodium leaks [20]. All mammalian cells maintain an ion gradient across their outer wall, which is utilized to fuel cell chemical processes. The so-called sodium pump is an active process that constantly pumps sodium out of the cell to maintain this charge difference. The pump consumes ATP in the process. Working against the pump is a passive leakage mechanism that causes sodium to drift back into the cell. To the extent that the membrane can be constructed to resist leakage (kind of like putting insulation in the attic of a house), it will require less ATP to maintain sodium concentrations appropriate for the cell to function properly.
The Haines article argues that cholesterol plays an essential role in protecting the cell wall from sodium leakage. Sodium leakage is a much bigger problem (it leaks 7 to 11 times as fast in the absence of cholesterol) for unsaturated fatty acids as for saturated fatty acids [4]. However, unsaturated fatty acids also encourage cholesterol to arrange itself in the central layer. By accumulating there, it provides extra insulation preventing the charged sodium ions from passively hopping from the exterior to the interior of the cell. Other experiments [30] have shown that the relative sodium leakage rates are reduced by 300% in the presence of cholesterol.
9. The Evidence of Statin Damage to Muscles
Typically in America, if a person fails a stress test or suffers from a heart attack and then is found to have a blocked coronary artery, a stent will be introduced to correct the problem and high-dose statin therapy will be initiated, with the expectation that the drug will be needed for the rest of their life. The accepted belief today is that, no matter whether their cholesterol is already low, high-dose statin therapy will yield sufficient benefit to offset any side effects it might cause. At the same time, these patients are encouraged to spend up to an hour a day exercising on a treadmill, since exercise has been shown to be highly beneficial to heart disease prognosis. The exercise, in conjunction with the metabolic deficiencies induced by the statin drug, are a potentially lethal combination.
Typically, also, the patient is not alerted that a common side effect of statin drugs is muscle pain and muscle weakness. It is often the case that such symptoms don't appear immediately. In fact, it can sometimes be years before statin therapy leads to enough damage to cause obvious symptoms. By that time, the person may well believe that the pain and weakness are simply a consequence of getting older.
It has been widely claimed, and statin users seem to have embraced this concept, that, as long as you monitor your enzyme levels, you can simply terminate statin therapy if the enzymes get too high, and all will be well. However, judging from some of the sad stories that are showing up in comment pages all over the web, this has turned out not to be the case for some people.
An article published in July, 2009 [27] investigated the association between physical muscle damage and patients' complaints of muscle weakness or pain. Patients who reported weakness said, for example, that it was difficult to get up from a seated position without arm support. Those who reported pain generally said that it was worse after physical exercise. Only one out of 44 patients examined developed overt rhabdomyolysis, with the serum level of the muscle enzyme creatine kinase measured at 57,657 U/L. This patient required hospital treatment for management of his pain.
The authors were interested in investigating the extent to which muscle damage could be seen through muscle biopsy for these patients. They compared them with 20 patients who had never taken a statin drug. Twenty five of the 44 patients taking statins had clear muscle damage. None of the 20 controls had any evidence of damage. Other than the one patient with overt rhabdomyolysis, none of the others had muscle enzyme levels above the cut-off considered the upper level of "normal." For those patients with injuries, on average 10% of their fibers were injured. The authors concluded that the lack of elevated levels of creatine kinase does not rule out structural muscle injury.
Typically, also, the patient is not alerted that a common side effect of statin drugs is muscle pain and muscle weakness. It is often the case that such symptoms don't appear immediately. In fact, it can sometimes be years before statin therapy leads to enough damage to cause obvious symptoms. By that time, the person may well believe that the pain and weakness are simply a consequence of getting older.
It has been widely claimed, and statin users seem to have embraced this concept, that, as long as you monitor your enzyme levels, you can simply terminate statin therapy if the enzymes get too high, and all will be well. However, judging from some of the sad stories that are showing up in comment pages all over the web, this has turned out not to be the case for some people.
An article published in July, 2009 [27] investigated the association between physical muscle damage and patients' complaints of muscle weakness or pain. Patients who reported weakness said, for example, that it was difficult to get up from a seated position without arm support. Those who reported pain generally said that it was worse after physical exercise. Only one out of 44 patients examined developed overt rhabdomyolysis, with the serum level of the muscle enzyme creatine kinase measured at 57,657 U/L. This patient required hospital treatment for management of his pain.
The authors were interested in investigating the extent to which muscle damage could be seen through muscle biopsy for these patients. They compared them with 20 patients who had never taken a statin drug. Twenty five of the 44 patients taking statins had clear muscle damage. None of the 20 controls had any evidence of damage. Other than the one patient with overt rhabdomyolysis, none of the others had muscle enzyme levels above the cut-off considered the upper level of "normal." For those patients with injuries, on average 10% of their fibers were injured. The authors concluded that the lack of elevated levels of creatine kinase does not rule out structural muscle injury.
10. Statins and Heart Failure
A paper titled simply "Lovastatin decreases coenzyme Q levels in humans" [16] states unequivocally in the abstract: "It is established that Coenzyme Q10 is indispensable for cardiac function." The heart is a muscle, and hence it is subject to all the same laws of physics as the skeletal muscles. It faces the same problem of fuel deficiency due to the various effects statins have on metabolism discussed above. Heart muscle cells would also have to cannibalize themselves to get enough fuel, and would also suffer damage to their cell membranes due to exposure to Ferryl myoglobin.
An article published in 2004 [42] provides a plausible theory for the process by which muscle cells in the heart become dysfunctional with old age, leading ultimately to heart failure. The argument blends perfectly with the logical deductions associated with the mechanism by which statins damage cells, and leads to the unavoidable conclusion that statins make you age at an accelerated pace. The process involves a downward spiral caused by deficiencies in both the mitochondria and the lysosomes. Recall that the mitochondria are responsible for providing fuel to the cell, and the lysosomes are responsible for digesting and decomposing residues of waste products. The article claims that the downward spiral is caused by "continuous physiological oxidative stress." Oxidative stress is greatly enhanced by statins, because they deplete the supply of both antioxidants like coenzyme Q10 and fresh phospholipids and cholesterol to rebuild damaged cell walls. Debris from damaged phospholipids in the cell wall, the mitochondrial walls, and the lysosome walls must be taken up by the lysosomes, digested, and disposed of. Under normal circumstances the lysosomes would easily break them down in their highly acidic environment, using their powerful digestive enzymes.
When the lysosomes are unable to digest the debris that accumulates from damaged cell walls, the residue that remains is called "lipofuscin." Lipofuscin is considered to be a signature of old age, accumulating in the liver, kidney, heart muscle, and nerve cells as we get older. Lipofuscin is believed to be the product of oxidation of unsaturated fatty acids, and is indicative of membrane damage, whether to the cell's outer wall or to the walls of the lysosomes and mitochondria [17].
For long-term statin users, lipofuscin almost certainly accumulates, because their lysosomes are dysfunctional. This condition would arise not just in the heart, but in all the cells of the body. As I mentioned earlier, statins cripple the production of the dolichols, antioxidants that play a crucial role in protecting the lysosomes from hydrogen ion leakage. Lysosomes also depend upon cholesterol in their membranes to provide additional insulation against charge dissipation. With a constant leakage outward of H+ ions, lysosomes can not maintain their pH at a sufficiently acidic level to allow their enzymes to work. As a consequence, undegradable debris, i.e., lipofuscin, accumulates within the lysosomes, and the cell has no backup repair system to salvage the disaster. The last sentence in the abstract of [42] says: "This interrelated mitochondrial and lysosomal damage eventually results in functional failure and death of cardiac myocytes {heart muscle cells]."
Doctor Peter Langsjoehn believes that statins are inducing an epidemic rise in the incidence of heart failure. He wrote: "In my practice of 17 years in Tyler, Texas, I have seen a frightening increase in heart failure secondary to statin usage, 'statin cardiomyopathy.' Over the past five years, statins have become more potent, are being prescribed in higher doses, and are being used with reckless abandon in the elderly and in patients with 'normal' cholesterol levels. We are in the midst of a CHF epidemic in the US with a dramatic increase over the past decade. Are we causing this epidemic through our zealous use of statins? In large part I think the answer is yes. " (Statins and Heart Failure) .
Dr. Duane Graveline, a long-time advocate of the dangers of statin therapy, has provided a very clear description ( Duane Graveline on Statins and Heart Failure) of the role of coenzyme Q10 in the heart and the reason why its inhibition by statins would lead to heart failure. You can find several references to relevant articles by Dr. Langsjoehn on that page.
A very recent study (November, 2009) [8] found that patients with diastolic heart failure who were taking statins had a significantly poorer outcome than patients who were not on statin therapy. Diastolic heart failure is distinguished from systolic heart failure in that it is associated with dysfunction of the heart during the resting phase rather than the contracting phase. However, it is the cause of nearly half of the cases of heart failure, and it is equally as fatal as the systolic form. In the study, it was confirmed that people with diastolic heart failure who were on statin therapy were more likely to have problems with their lungs and were less able to exert themselves (weaker muscles, poorer exercise tolerance) than those not on statins.
An article published in 2004 [42] provides a plausible theory for the process by which muscle cells in the heart become dysfunctional with old age, leading ultimately to heart failure. The argument blends perfectly with the logical deductions associated with the mechanism by which statins damage cells, and leads to the unavoidable conclusion that statins make you age at an accelerated pace. The process involves a downward spiral caused by deficiencies in both the mitochondria and the lysosomes. Recall that the mitochondria are responsible for providing fuel to the cell, and the lysosomes are responsible for digesting and decomposing residues of waste products. The article claims that the downward spiral is caused by "continuous physiological oxidative stress." Oxidative stress is greatly enhanced by statins, because they deplete the supply of both antioxidants like coenzyme Q10 and fresh phospholipids and cholesterol to rebuild damaged cell walls. Debris from damaged phospholipids in the cell wall, the mitochondrial walls, and the lysosome walls must be taken up by the lysosomes, digested, and disposed of. Under normal circumstances the lysosomes would easily break them down in their highly acidic environment, using their powerful digestive enzymes.
When the lysosomes are unable to digest the debris that accumulates from damaged cell walls, the residue that remains is called "lipofuscin." Lipofuscin is considered to be a signature of old age, accumulating in the liver, kidney, heart muscle, and nerve cells as we get older. Lipofuscin is believed to be the product of oxidation of unsaturated fatty acids, and is indicative of membrane damage, whether to the cell's outer wall or to the walls of the lysosomes and mitochondria [17].
For long-term statin users, lipofuscin almost certainly accumulates, because their lysosomes are dysfunctional. This condition would arise not just in the heart, but in all the cells of the body. As I mentioned earlier, statins cripple the production of the dolichols, antioxidants that play a crucial role in protecting the lysosomes from hydrogen ion leakage. Lysosomes also depend upon cholesterol in their membranes to provide additional insulation against charge dissipation. With a constant leakage outward of H+ ions, lysosomes can not maintain their pH at a sufficiently acidic level to allow their enzymes to work. As a consequence, undegradable debris, i.e., lipofuscin, accumulates within the lysosomes, and the cell has no backup repair system to salvage the disaster. The last sentence in the abstract of [42] says: "This interrelated mitochondrial and lysosomal damage eventually results in functional failure and death of cardiac myocytes {heart muscle cells]."
Doctor Peter Langsjoehn believes that statins are inducing an epidemic rise in the incidence of heart failure. He wrote: "In my practice of 17 years in Tyler, Texas, I have seen a frightening increase in heart failure secondary to statin usage, 'statin cardiomyopathy.' Over the past five years, statins have become more potent, are being prescribed in higher doses, and are being used with reckless abandon in the elderly and in patients with 'normal' cholesterol levels. We are in the midst of a CHF epidemic in the US with a dramatic increase over the past decade. Are we causing this epidemic through our zealous use of statins? In large part I think the answer is yes. " (Statins and Heart Failure) .
Dr. Duane Graveline, a long-time advocate of the dangers of statin therapy, has provided a very clear description ( Duane Graveline on Statins and Heart Failure) of the role of coenzyme Q10 in the heart and the reason why its inhibition by statins would lead to heart failure. You can find several references to relevant articles by Dr. Langsjoehn on that page.
A very recent study (November, 2009) [8] found that patients with diastolic heart failure who were taking statins had a significantly poorer outcome than patients who were not on statin therapy. Diastolic heart failure is distinguished from systolic heart failure in that it is associated with dysfunction of the heart during the resting phase rather than the contracting phase. However, it is the cause of nearly half of the cases of heart failure, and it is equally as fatal as the systolic form. In the study, it was confirmed that people with diastolic heart failure who were on statin therapy were more likely to have problems with their lungs and were less able to exert themselves (weaker muscles, poorer exercise tolerance) than those not on statins.
11. Statins and Lung Disease
The statin industry has tried to promote the idea that statins might be beneficial in treating pneumonia. They came to this erroneous conclusion through retrospective studies, where the observed benefits come, I suspect, from the fact that those who took statins had benefited from high cholesterol for probably many years before introducing statin therapy. The industry was sufficiently encouraged by preliminary positive indications to then conduct placebo-controlled studies to try to legitimize their claim. However, the studies backfired, because they clearly showed that statin therapy not only wasn't helpful, but actually led to a significantly worse prognosis [26][12] (see ( Statins Increase Pneumonia Risk) . For pneumonia severe enough to require hospitalization, the increased risk incurred by taking a statin was an alarming 61% [12].
Statins' effects on muscles apply to the respiratory muscles as well, leading to difficulty in breathing and subsequent oxygen deprivation, which, of course, further aggravates both pneumonia and heart failure. Furthermore, it is now well known that, in rare cases, statin drugs cause severe lung disease, so-called "interstitial lung disease" (ILD) [24][44][15]. ILD is now listed as a rare side effect for all statin drugs.
In an excellent review article published in 2008, Fernandez et al. [15] identify several possibilities for how statins might cause interstitial pneumonia. They begin their discussion by drawing an analogy with amiodarone, a drug which is known to cause a very similar kind of pathology, which includes the accumulation of lysosomal inclusion bodies, i.e., lipofuscin, the cell-membrane debris that was described previously in the section under heart disease.
Amiodarone belongs to a very common class of drugs known as "amphiphilic" drugs: they have both a hydrophilic (water soluble) and a lipophilic (fat soluble) component in their chemical structure. This property allows them to cross through the membranes of cells in order to achieve their desired biochemical influence. However, the process by which they enter the cell involves degrading the lipids in the cell membrane [2] . Membrane fragments break away from the cell wall and carry the drug along with them into the cell. As a consequence of cell wall deterioration, sodium leaks will cause the cell to lose energy, with all the negative consequences that have been described before.
Fernandez et al. argue that, like amiodarone, statins have an amphiphilic structure, since they contain an apolar (lipophilic) ring and a hydrophilic side chain. A really disturbing observation they make is that, over time, amphiphilic drugs are known to become more efficient at entering cells. It seems logical that a deteriorating cell wall would allow better permeability to the drug molecule. But this then means that whatever effect the drug has on the cell will be increased, leading to accelerated damage and a destructive cascade.
Amiodarone is a potent antidysrhythmic agent, i.e., a drug used to try to correct an irregular heart beat during heart failure or post-operative. It has numerous side effects, but probably the most serious side effect is interstitial lung disease. An article written in 2001 [3] explored the likely mechanism of pulmonary damage. The authors conducted in vitro experiments on cells in lung tissue extracted from hamsters. They noted that exposure to the drug decreased the mitochondrial membrane potential (H+ ions leaked out of the mitochondria) and subsequently the amount of ATP in the cell dropped by 32 to 77%. Even with the addition of glucose, the mitochondria were not able to regenerate the depleted ATP; i.e., the mitochondria were not functioning properly to generate energy from glucose. Ultimately, the cells died. They concluded that mitochondrial dysfunction was the path by which the drug induced cell death.
What they describe is essentially the exact same process by which statins lead to problems in muscle cells. Fernandez et al. agree with my argument that, like amiodarone, statins may cause interstitial lung disease through their disruption of the mitochondrial electron transport chain and subsequent depletion of ATP. Lung cells are particularly vulnerable to oxidative damage, because they are tasked with capturing oxygen from the air and transporting it to the blood. I also suspect that, although the number of cases of reported interstial disease is small, there is a much larger number of people whose lungs have been compromised by statins, but whose pulmonary function has not yet deteriorated to a catastrophic point. Instead, they experience some difficulty breathing and a perceived inability to get enough oxygen. As with muscle weakness, such symptoms may go unreported, as the patient has no way of knowing that what he is experiencing is not a normal aspect of growing old. Certainly an increased susceptibility to viral pneumonia would be anticipated when the lung's cells are suffering from insufficient energy and a degraded cell wall.
Statins' effects on muscles apply to the respiratory muscles as well, leading to difficulty in breathing and subsequent oxygen deprivation, which, of course, further aggravates both pneumonia and heart failure. Furthermore, it is now well known that, in rare cases, statin drugs cause severe lung disease, so-called "interstitial lung disease" (ILD) [24][44][15]. ILD is now listed as a rare side effect for all statin drugs.
In an excellent review article published in 2008, Fernandez et al. [15] identify several possibilities for how statins might cause interstitial pneumonia. They begin their discussion by drawing an analogy with amiodarone, a drug which is known to cause a very similar kind of pathology, which includes the accumulation of lysosomal inclusion bodies, i.e., lipofuscin, the cell-membrane debris that was described previously in the section under heart disease.
Amiodarone belongs to a very common class of drugs known as "amphiphilic" drugs: they have both a hydrophilic (water soluble) and a lipophilic (fat soluble) component in their chemical structure. This property allows them to cross through the membranes of cells in order to achieve their desired biochemical influence. However, the process by which they enter the cell involves degrading the lipids in the cell membrane [2] . Membrane fragments break away from the cell wall and carry the drug along with them into the cell. As a consequence of cell wall deterioration, sodium leaks will cause the cell to lose energy, with all the negative consequences that have been described before.
Fernandez et al. argue that, like amiodarone, statins have an amphiphilic structure, since they contain an apolar (lipophilic) ring and a hydrophilic side chain. A really disturbing observation they make is that, over time, amphiphilic drugs are known to become more efficient at entering cells. It seems logical that a deteriorating cell wall would allow better permeability to the drug molecule. But this then means that whatever effect the drug has on the cell will be increased, leading to accelerated damage and a destructive cascade.
Amiodarone is a potent antidysrhythmic agent, i.e., a drug used to try to correct an irregular heart beat during heart failure or post-operative. It has numerous side effects, but probably the most serious side effect is interstitial lung disease. An article written in 2001 [3] explored the likely mechanism of pulmonary damage. The authors conducted in vitro experiments on cells in lung tissue extracted from hamsters. They noted that exposure to the drug decreased the mitochondrial membrane potential (H+ ions leaked out of the mitochondria) and subsequently the amount of ATP in the cell dropped by 32 to 77%. Even with the addition of glucose, the mitochondria were not able to regenerate the depleted ATP; i.e., the mitochondria were not functioning properly to generate energy from glucose. Ultimately, the cells died. They concluded that mitochondrial dysfunction was the path by which the drug induced cell death.
What they describe is essentially the exact same process by which statins lead to problems in muscle cells. Fernandez et al. agree with my argument that, like amiodarone, statins may cause interstitial lung disease through their disruption of the mitochondrial electron transport chain and subsequent depletion of ATP. Lung cells are particularly vulnerable to oxidative damage, because they are tasked with capturing oxygen from the air and transporting it to the blood. I also suspect that, although the number of cases of reported interstial disease is small, there is a much larger number of people whose lungs have been compromised by statins, but whose pulmonary function has not yet deteriorated to a catastrophic point. Instead, they experience some difficulty breathing and a perceived inability to get enough oxygen. As with muscle weakness, such symptoms may go unreported, as the patient has no way of knowing that what he is experiencing is not a normal aspect of growing old. Certainly an increased susceptibility to viral pneumonia would be anticipated when the lung's cells are suffering from insufficient energy and a degraded cell wall.
12. Statins and Diabetes
The JUPITER trial of the statin drug Crestor was widely heralded as evidence that statin drugs can delay heart attacks for people who have high levels of an indicator of inflammation called C-reactive protein. However, what is less known about this trial is that it uncovered a clear link between statin drugs (or, at least, Crestor) and increased risk of diabetes (JUPITER Trial and Diabetes) [36]. According to Dr. Jay Cohen, the people who took Crestor in the trial had a 25% increased risk of developing diabetes, compared to the control group. This is alarming, because diabetes itself is an extremely strong risk factor for heart disease.
The pancreas synthesizes insulin in its beta cells, and defects in insulin production (either too little of it, or a lack of response to it) is the cause of diabetes. Insulin is used by the body's cells to catalyze the transport of glucose into the cell. Without insulin, or with poorly functioning insulin, sugar piles up in the blood and the cells become energy starved.
There have been a large number of studies on the biochemistry of the beta cells and their insulin-producing machinery, and it has been determined that beta cells require both cholesterol [46] and fats [11] to be present before they will release insulin. Inadequate cholesterol and poor quality phospholipids in the beta cell's outer membrane likely impair its ability to transport insulin across the membrane. Statin drugs, of course, reduce the bioavailability of cholesterol, but also of fatty acids, because these are transported in the blood stream via the same LDL particles that statins suppress. Thus, it is easy to see why statins would cause an increased risk to diabetes.
In addition to the above defects in the cell membrane, impaired function of the mitochondria in the beta cells has also been clearly implicated in diabetes, in studies involving diabetic mice with defective mitochondrial genes [39]. These mice exhibited reduced insulin secretion when they were only five weeks old, and their mitochondria were abnormal in appearance and were unable to maintain an adequate charge gradient across their membranes. In other words, they exhibited defects that are similar to what would be expected with reduced coenzyme Q10 as a consequence of statin exposure. Older mice with the same defect were severely deficient in insulin production, as many of their pancreatic beta cells had died off.
Insulin suppresses the release of fats from both the fat cells and the liver, and therefore there will be a fat shortage in the blood supply subsequent to insulin release, unless abundant fats are already present. Thus, it is a good strategy, biologically, for the beta cells to be sure fats and cholesterol are well supplied before injecting insulin into the blood stream. I have previously written extensively on this subject (Essay on Metabolic Syndrome).
A study published in March, 2009 [41] looked at the relationship between statin drug usage and fasting blood glucose levels, the test typically conducted to assess diabetes risk. They grouped 345,417 patients into two categories: with or without a previous diabetes diagnosis. They compared fasting glucose levels before they began taking statins and then after they had been on statins for an average of two years. In both groups, they obtained a highly significant (P < 0.0001) result of increased fasting glucose levels for those on statin therapy.
A reduction in the ability of glucose to enter muscle cells, consequential to a reduction in insulin supply, would add insult onto injury for the muscle cells trying to survive with a defective aerobic metabolism factory. Because the muscles are forced to switch to the much less efficient anaerobic metabolism of glucose in order to avoid oxidative damage, they require enormously more glucose to meet their energy supply than they would require if their mitochondrial energy-generating factory were functioning properly. Yet the reduced insulin is making it harder to get enough glucose in. This will force the cell into the starvation mode that leads to cannibalization of its internal muscle protein. The perceived result over time will be extreme muscle weakness.
The pancreas synthesizes insulin in its beta cells, and defects in insulin production (either too little of it, or a lack of response to it) is the cause of diabetes. Insulin is used by the body's cells to catalyze the transport of glucose into the cell. Without insulin, or with poorly functioning insulin, sugar piles up in the blood and the cells become energy starved.
There have been a large number of studies on the biochemistry of the beta cells and their insulin-producing machinery, and it has been determined that beta cells require both cholesterol [46] and fats [11] to be present before they will release insulin. Inadequate cholesterol and poor quality phospholipids in the beta cell's outer membrane likely impair its ability to transport insulin across the membrane. Statin drugs, of course, reduce the bioavailability of cholesterol, but also of fatty acids, because these are transported in the blood stream via the same LDL particles that statins suppress. Thus, it is easy to see why statins would cause an increased risk to diabetes.
In addition to the above defects in the cell membrane, impaired function of the mitochondria in the beta cells has also been clearly implicated in diabetes, in studies involving diabetic mice with defective mitochondrial genes [39]. These mice exhibited reduced insulin secretion when they were only five weeks old, and their mitochondria were abnormal in appearance and were unable to maintain an adequate charge gradient across their membranes. In other words, they exhibited defects that are similar to what would be expected with reduced coenzyme Q10 as a consequence of statin exposure. Older mice with the same defect were severely deficient in insulin production, as many of their pancreatic beta cells had died off.
Insulin suppresses the release of fats from both the fat cells and the liver, and therefore there will be a fat shortage in the blood supply subsequent to insulin release, unless abundant fats are already present. Thus, it is a good strategy, biologically, for the beta cells to be sure fats and cholesterol are well supplied before injecting insulin into the blood stream. I have previously written extensively on this subject (Essay on Metabolic Syndrome).
A study published in March, 2009 [41] looked at the relationship between statin drug usage and fasting blood glucose levels, the test typically conducted to assess diabetes risk. They grouped 345,417 patients into two categories: with or without a previous diabetes diagnosis. They compared fasting glucose levels before they began taking statins and then after they had been on statins for an average of two years. In both groups, they obtained a highly significant (P < 0.0001) result of increased fasting glucose levels for those on statin therapy.
A reduction in the ability of glucose to enter muscle cells, consequential to a reduction in insulin supply, would add insult onto injury for the muscle cells trying to survive with a defective aerobic metabolism factory. Because the muscles are forced to switch to the much less efficient anaerobic metabolism of glucose in order to avoid oxidative damage, they require enormously more glucose to meet their energy supply than they would require if their mitochondrial energy-generating factory were functioning properly. Yet the reduced insulin is making it harder to get enough glucose in. This will force the cell into the starvation mode that leads to cannibalization of its internal muscle protein. The perceived result over time will be extreme muscle weakness.
13. Statins and Muscle Damage: Conclusions
If you live in the United States, and your doctor has identified that you are at high risk to heart attacks, he has likely prescribed a high dose statin even if your cholesterol levels are not high. You have likely also been put on a low-fat, low saturated fat diet, and you have been encouraged to work out on a treadmill every day.
My research indicates that, if you rigorously follow all of your doctor's advice, you will be facing severe muscle damage sooner or later. The statin drug's impact on the mitochondria and on the cell walls of the muscle cells is such that even modest exercise can lead to rhabdomyolysis. For some it will be obvious right away that the side effects are too damaging and the statin therapy must be terminated. For others, the damage will happen more insidiously, and will not become apparent until years after statin therapy was initiated. But often patients will find that the symptoms remain after the drug is stopped -- it will be too late to repair the muscle damage. Or, worse, they will develop kidney failure or heart failure as a consequence.
Statin drugs have many adverse side effects, but probably the most frequent complaints concern muscle pain and muscle weakness. In this essay, I have developed a physiological explanation for the mechanism responsible for this side effect. It is due to the fact that statins interfere with the synthesis of not only cholesterol, but also coenzyme Q10 and the dolichols. Statins also reduce the bioavailability to the cells of both fatty acids and all dietary antioxidants, due to the sharp reduction in serum levels of LDL, which delivers these essential nutrients to the cells.
Without sufficient coenzyme Q10, muscle cells suffer from an impaired ability to generate energy to fuel their contractions. They are forced to cannibalize their own proteins to survive. At the same time, powerful oxidative agents are generated which damage the myoglobin in the cell, rendering it both ineffective to transport oxygen and toxic to the cell wall. The oxidized myoglobin, known as "Ferryl myoglobin" is toxic to the fatty acids that are the main component of the cell wall. With insufficient cholesterol in the cell wall, the cell can't hold a charge, and this also causes it to waste energy. The lysosomes are unable to digest debris because they can't maintain a sufficiently acidic environment. The problem is further compounded by profound shortages of cholesterol, which would have offered further protection against oxidative damage to the fatty acids and ion leakage in the cell wall, the mitochondrial wall, and the lysosome wall. Eventually the cell disintegrates and the myoglobin is released into the blood stream. It makes its way to the kidneys, which try to dispose of it. But the Ferryl myoglobin is also toxic to the kidneys, which leads to severe kidney disease.
The low-fat diet and the exercise regime will both increase the likelihood that the statin drug will cause problems. Vigorous exercise increases the energy needs of the muscles, while the low-fat diet reduces even further the bioavailability of fatty acids to replace damaged cell walls. Furthermore, cell walls composed of unsaturated fats are more vulnerable to attack by the Ferryl myoglobin than those composed of saturated fats.
Because the heart is also a muscle, it also suffers from damage due to exposure to statins. This leads to a reduced likelihood of recovering from a diastolic heart attack, and an increased chance of developing heart failure. Damaged cells of the respiratory system lead to an increased risk of both pneumonia and interstitial lung disease, both of which are very dangerous for someone with a weak heart.
The JUPITER trial revealed that the treatment group had a 25% increased risk for diabetes, and I have explained above why this would be true. Diabetes is a significant risk factor for heart disease, so this outcome is disturbing, and one wonders whether the trial was terminated early to avoid making this number even worse. Dr. William Davis, a cardiologist who believes that statins should be a last resort in treating heart disease, has this to say about the JUPITER trial: "I view the foisting of Crestor via the JUPITER argument on the public as taking full advantage of the helpless situation many Americans find themselves in: Reduce fat intake, eat more healthy whole grains and . . . cholesterol and CRP skyrocket! 'You need Crestor! See, I told you it was genetic,' says the doctor after attending the nice AstraZeneca-sponsored drug dinner." ( Dr. Davis' Blog Post on JUPITER)
The news has just come out that even children are now being tested for high cholesterol, and it is being suggested that they should be put on a statin drug if they can not control their cholesterol levels (Children Taking Statins??). I find this news to be extremely disturbing, especially since none of the controlled statin trials have been conducted on children. We have no idea what negative consequences statin drugs might have on the developing nervous system of a child. However, it has been shown that statins can completely destroy the nervous system of an embryo [13].
A remarkable recent publication by Jeff Cable (December, 2009) [7] analyzes a set of 885 self-reported adverse effects of statin therapy by patients. Although the reports covered a wide range of known side effects of statins, including cognitive impairment, muscle pain and weakness, skin problems and sexual dysfunction, what was most disturbing was the large number of reports of severe neurological damage. Most distressing was the fact that there were a total of 17 reports of ALS with 2 additional reports related to motor neuron deterioration, which he counts together as 1 to give a total of 18. In ALS, nerve cells waste away or die, and can no longer send messages to muscles. This eventually leads to muscle weakening, twitching, and ultimately paralysis. As the disease progresses, swallowing and breathing become difficult. Most victims die within five years of diagnosis.
The author's comments related to neurological disorders and ALS are quoted here: "One fragment of information that was gained from the patient accounts is the apparent incidence of major neurodegenerative diseases which may well have been precipitated by statin therapy. ... The rarest of these conditions is ALS and yet in just 351 reports there were enough cases to have made the prediction (based upon incidence statistics) that an expected three million six hundred thousand accounts would have to be written before eighteen ALS/MND cases would have been revealed. This is such an astonishingly high number of cases to report within such a small participant group that it would be right to ask whether a fundamental error has been made. Absent any error it is also right to ask: What is really happening? What is the real risk posed by statin therapy?"
There is prior evidence from the literature implicating a relationship between statins and ALS -- a study of the FDA's adverse event reports [10] as well as a study showing that high cholesterol protects against ALS [19]. My next essay will be on the subject of statin drugs' likely adverse effects on the nervous system: I will argue that statins increase risk not just to ALS but to multiple sclerosis, Parkinson's disease, and Alzheimer's.
My research indicates that, if you rigorously follow all of your doctor's advice, you will be facing severe muscle damage sooner or later. The statin drug's impact on the mitochondria and on the cell walls of the muscle cells is such that even modest exercise can lead to rhabdomyolysis. For some it will be obvious right away that the side effects are too damaging and the statin therapy must be terminated. For others, the damage will happen more insidiously, and will not become apparent until years after statin therapy was initiated. But often patients will find that the symptoms remain after the drug is stopped -- it will be too late to repair the muscle damage. Or, worse, they will develop kidney failure or heart failure as a consequence.
Statin drugs have many adverse side effects, but probably the most frequent complaints concern muscle pain and muscle weakness. In this essay, I have developed a physiological explanation for the mechanism responsible for this side effect. It is due to the fact that statins interfere with the synthesis of not only cholesterol, but also coenzyme Q10 and the dolichols. Statins also reduce the bioavailability to the cells of both fatty acids and all dietary antioxidants, due to the sharp reduction in serum levels of LDL, which delivers these essential nutrients to the cells.
Without sufficient coenzyme Q10, muscle cells suffer from an impaired ability to generate energy to fuel their contractions. They are forced to cannibalize their own proteins to survive. At the same time, powerful oxidative agents are generated which damage the myoglobin in the cell, rendering it both ineffective to transport oxygen and toxic to the cell wall. The oxidized myoglobin, known as "Ferryl myoglobin" is toxic to the fatty acids that are the main component of the cell wall. With insufficient cholesterol in the cell wall, the cell can't hold a charge, and this also causes it to waste energy. The lysosomes are unable to digest debris because they can't maintain a sufficiently acidic environment. The problem is further compounded by profound shortages of cholesterol, which would have offered further protection against oxidative damage to the fatty acids and ion leakage in the cell wall, the mitochondrial wall, and the lysosome wall. Eventually the cell disintegrates and the myoglobin is released into the blood stream. It makes its way to the kidneys, which try to dispose of it. But the Ferryl myoglobin is also toxic to the kidneys, which leads to severe kidney disease.
The low-fat diet and the exercise regime will both increase the likelihood that the statin drug will cause problems. Vigorous exercise increases the energy needs of the muscles, while the low-fat diet reduces even further the bioavailability of fatty acids to replace damaged cell walls. Furthermore, cell walls composed of unsaturated fats are more vulnerable to attack by the Ferryl myoglobin than those composed of saturated fats.
Because the heart is also a muscle, it also suffers from damage due to exposure to statins. This leads to a reduced likelihood of recovering from a diastolic heart attack, and an increased chance of developing heart failure. Damaged cells of the respiratory system lead to an increased risk of both pneumonia and interstitial lung disease, both of which are very dangerous for someone with a weak heart.
The JUPITER trial revealed that the treatment group had a 25% increased risk for diabetes, and I have explained above why this would be true. Diabetes is a significant risk factor for heart disease, so this outcome is disturbing, and one wonders whether the trial was terminated early to avoid making this number even worse. Dr. William Davis, a cardiologist who believes that statins should be a last resort in treating heart disease, has this to say about the JUPITER trial: "I view the foisting of Crestor via the JUPITER argument on the public as taking full advantage of the helpless situation many Americans find themselves in: Reduce fat intake, eat more healthy whole grains and . . . cholesterol and CRP skyrocket! 'You need Crestor! See, I told you it was genetic,' says the doctor after attending the nice AstraZeneca-sponsored drug dinner." ( Dr. Davis' Blog Post on JUPITER)
The news has just come out that even children are now being tested for high cholesterol, and it is being suggested that they should be put on a statin drug if they can not control their cholesterol levels (Children Taking Statins??). I find this news to be extremely disturbing, especially since none of the controlled statin trials have been conducted on children. We have no idea what negative consequences statin drugs might have on the developing nervous system of a child. However, it has been shown that statins can completely destroy the nervous system of an embryo [13].
A remarkable recent publication by Jeff Cable (December, 2009) [7] analyzes a set of 885 self-reported adverse effects of statin therapy by patients. Although the reports covered a wide range of known side effects of statins, including cognitive impairment, muscle pain and weakness, skin problems and sexual dysfunction, what was most disturbing was the large number of reports of severe neurological damage. Most distressing was the fact that there were a total of 17 reports of ALS with 2 additional reports related to motor neuron deterioration, which he counts together as 1 to give a total of 18. In ALS, nerve cells waste away or die, and can no longer send messages to muscles. This eventually leads to muscle weakening, twitching, and ultimately paralysis. As the disease progresses, swallowing and breathing become difficult. Most victims die within five years of diagnosis.
The author's comments related to neurological disorders and ALS are quoted here: "One fragment of information that was gained from the patient accounts is the apparent incidence of major neurodegenerative diseases which may well have been precipitated by statin therapy. ... The rarest of these conditions is ALS and yet in just 351 reports there were enough cases to have made the prediction (based upon incidence statistics) that an expected three million six hundred thousand accounts would have to be written before eighteen ALS/MND cases would have been revealed. This is such an astonishingly high number of cases to report within such a small participant group that it would be right to ask whether a fundamental error has been made. Absent any error it is also right to ask: What is really happening? What is the real risk posed by statin therapy?"
There is prior evidence from the literature implicating a relationship between statins and ALS -- a study of the FDA's adverse event reports [10] as well as a study showing that high cholesterol protects against ALS [19]. My next essay will be on the subject of statin drugs' likely adverse effects on the nervous system: I will argue that statins increase risk not just to ALS but to multiple sclerosis, Parkinson's disease, and Alzheimer's.
Acknowledgements for Essay on Statins and Muscle Damage
I would like to thank Glyn Wainwright for pointing me to both his own excellent review paper and the very informative and fascinating article by Haines [20] on proton and sodium leaks through lipid bilayers, which played a crucial role in my arguments for statin damage to muscles.
References for Essay on Statins and Muscle Damage
[1] A Arduini, L. Eddy, and P. Hochstein, "Detection of ferryl myoglobin in the isolated ischemic rat heart," Free-Radic-Biol-Med. (1990) Vol. 9, No. 6, pp. 511-3.
[2] M. Baciu, S.C. Sebai, O. Ces, X. Mulet, J.A. Clarke, G.C. Shearman, and R.V. Law, Templer RH, Plisson C, Parker CA, Gee A. "Degradative transport of cationic amphiphilic drugs across phospholipid bilayers." Philos Transact A Math Phys Eng Sci. (2006) Oct 15, Vol. 364(1847), pp. 2597-614.
[3] M.W. Bolt,J. W. Card, W.J. Racz, J.F. Brien and T.E. Massey, "Disruption of Mitochondrial Function and Cellular ATP Levels by Amiodarone and N-Desethylamiodarone in Initiation of Amiodarone-Induced Pulmonary Cytotoxicity," JPET (2001) September 1, Vol. 298, No. 3, pp. 1280-1289.
[4] S.L. Bonting, P.J. van Breugel, F.J. Daemen, "Influence of the lipid environment of the properties of rhodopsin in the photoreceptor membrane," Adv. Exp. Med. Biol. (1977) Vol. 83, pp. 175-89.
[5] P. Brancaccio, N. Maffulli, and F.M. Limongelli, "Creatine kinase monitoring in sport medicine" British Medical Bulletin (2007) Vol. 81-82. No. 1, pp. 209-230; doi:10.1093/bmb/ldm014
[6] E. Bruckert, G. Hayem, S. Dejager, et al. "Mild to moderate muscular symptoms with high-dosage statin therapy in hyperlipidemic patients -- the PRIMO study." Cardiovasc Drugs Ther (2005) Vol. 19, pp. 403-14.
[7] J. Cable, "Adverse Events of Statins -- An Informal Internet-based Study," JOIMR (2009, December, Vol. 7, No. 1; http://www.joimr.org/JOIMR_Vol7_No1_Dec2009.pdf.
[8] L.P. Cahalin, PT, PhD, et al., CHEST 2009: American College of Chest Physicians Annual Meeting, Poster 592. Presented November 4, 2009.
[9] J.C. Chatham, "Lactate - the forgotten fuel!" J Physiol. (2002) July 15; 542(Pt 2), p. 333. doi: 10.1113/jphysiol.2002.020974.
[10] E. Colman, A. Szarfman, J. Wyeth, et al., "An evaluation of a data mining signal for amyotrophic lateral sclerosis and statins detected in FDA"s spontaneous adverse event reporting system," Pharmacoepidemiol Drug Saf (2008) Vol. 17, pp. 1060-76.
[11] B.E. Corkey, J.T. Deeney, G.C. Yaney, K. Tornheim, and M. Prentki, "The Role of Long-Chain Fatty Acyl-CoA Esters in Beta-Cell Signal Transduction," American Society for Nutritional Sciences, (2000) pp. 299S-304S.
[12] S. Dublin, M.L. Jackson, J.C. Nelson, N.S. Weiss, E.B. Larson, and L.A. Jackson, "Statin use and risk of community acquired pneumonia in older people: population based case-control study," BMJ (2009) Vol. 338, p. b2137 ; doi:10.1136/bmj.b2137
[13] R.J. Edison and M. Muenke, "Central nervous system and limb anomalies in case reports of first-trimester statin exposure," N Engl J Med (2004) Vol. 350, pp. 1579-1582.
[14] J.L. Farber, "Mechanisms of cell injury by activated oxygen species." Environ Health Perspect. (1994) December; Vol. 102 (Suppl 10), pp. 17-24.
[15] A.B. Fernandez, R.H. Karas, A.A. Alsheikh-Ali, and P.D. Thompson, "Statins and interstitial lung disease: a systematic review of the literature and of food and drug administration adverse event reports." Chest, (2008) Oct, Vol. 134 No. 4, pp. 824-30. Epub 2008 Aug 8.
[16] K. Folkers, P. Langsjoen, R. Willis, P. Richardson,L.J. Xia,C.Q. Ye, and H. Tamagawa, "Lovastatin decreases coenzyme Q levels in humans," PNAS (1990) November 1, Vol. 87, No. 22, pp. 8931-8934.
[17] C. Gaugler, "Lipofuscin", Stanislaus Journal of Biochemical Reviews
May (1997).
[18] Ghirlanda G, Oradei A, Manto A, Lippa S, Uccioli L, Caputo S, Greco A, Littarru G (1993). "Evidence of plasma CoQ10-lowering effect by HMG-CoA reductase inhibitors: a double-blind, placebo-controlled study". J Clin Pharmacol 33 (3): 226-9. PMID 8463436.
[19] M.R. Goldstein, L. Mascitelli, and F. Pezzetta, "Dyslipidemia is a protective factor in amyotrophic lateral sclerosis," Neurology (2008) Vol. 71, p. 956.
[20] T. H. Haines, "Do Sterols Reduce Proton and Sodium Leaks through Lipid Bilayers?" Progress in Lipid Research (2001) Vol.40, pp. 299-324.
[21] S. Jamil and P. Iqbal, "Rhabdomyolysis induced by a single dose of a statin." Heart (2004) Jan; Vol. 90 No. 1, p. e3.
[22] N. Kucerka, D. Marquardt, T.A. Harroun, M-P Nieh, S. R. Wassall, and J. Katsaras, "The Functional Significance of Lipid Diversity: Orientation of Cholesterol in Bilayers is Determined by Lipid Species," J. Am. Chem. Soc. (2009) Vol. 131, pp. 16358-16359.
[23] J.M. Land, J.A. Morgan-Hughes, and J.B.Clark, " Mitochondrial myopathy: biochemical studies revealing a deficiency of NADH-cytochrome b reductase activity." J. Neurol. Sci. 50: 1-13, 1981.
[24] S. Lantuejoul, E. Brambilla, C. Brambilla, and G. Devouassoux, "Statin-induced Fibrotic Nonspecific Interstitial Pneumonia," Eur Respir J. (2002) Vol. 19, pp. 577-580.
[25] R.S. Lees and A.M. Lees, "Rhabdomyolysis from the Coadministration of Lovastatin and the Antifungal Agent Itraconazole," NEJM (1995) Vol. 333, pp. 664-665.
[26] S.R. Majumdar, F.A. McAlister, D.T. Eurich, R.S. Padwal, and T.J. Marrie, "Statins and outcomes in patients admitted to hospital with community acquired pneumonia: population based prospective cohort study," BMJ (2006) Vol. 333, p. 999.
[27] M.G. Mohaupt, MD, R.H. Karas, MD PhD, E.B. Babiychuk, PhD, V. Sanchez-Freire, K. Monastyrskaya, PhD, L. Iyer, PhD, H. Hoppeler, MD, F. Breil and A. Draeger, MD "Association between statin-associated myopathy and skeletal muscle damage," CMAJ (2009) July 7 Vol. 181 No. 1-2 ; doi:10.1503/cmaj.081785.
[28] A. Mordente, S. A. Santini, G. A. D. Miggiano, G. E. Martorana, T. Petitti, G. Minotti, and B. Giardina, "The Interaction of Short Chain Coenzyme Q analogs with Different Redox States of Myoglobin," The Journal of Biological Chemistry, (1994) Vol. 269, Mo. 44, pp. 27394-27400.
[29] R.A. Oleka, J. Antosiewicza, J. Popinigisa, R. Gabbianellib, D. Fedelib and G. Falcionib, "Pyruvate but not lactate prevents NADH-induced myoglobin oxidation," Free Radical Biology and Medicine (2005) June; Vol. 38, Issue 11, pp. 1484-1490; doi:10.1016/j.freeradbiomed.2005.02.018.
[30] D. Papahadjopoulosa "Na+-K+ discrimination by “pure” phospholipid membranes," Biochimica et Biophysica Acta (BBA) (1971) Vol. 241, Issue 1, 6 July 1971, pp. 254-259
[31] R.P. Patel, U. Diczfalusy, S. Dzeletovic, M.T. Wilson and V.M. Darley-Usmar, "Formation of oxysterols during oxidation of low density lipoprotein by peroxynitrite, myoglobin, and copper, "Journal of Lipid Research (1996) Vol. 37, pp. 2361-2371.
[32] S. Pitkanen, A. Feigenbaum,, R. Laframboise, and B.H. Robinson, "NADH-coenzyme Q reductase (complex I) deficiency: heterogeneity in phenotype and biochemical findings," J. Inherit. Metab. Dis. (1996) Vol. 19, pp. 675-686.
[33] E.Y. Plotnikov, A.A. Chupyrkina, I.B. Pevzner, N.K. Isaev, and D.B. Zorov, "Myoglobin causes oxidative stress, increase of NO production and dysfunction of kidney's mitochondria," Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease (2009) Vol. 1792, Issue 8, August pp. 796-803; doi:10.1016/j.bbadis.2009.06.005
[34] T. Pozefsky, R G Tancredi, R T Moxley, J Dupre, and J D Tobin "Effects of brief starvation on muscle amino acid metabolism in nonobese man." J Clin Invest. (1976) February, Vol. 57, No. 2, pp. 444-449. doi: 10.1172/JCI108295.
[35] S.I. Rao, A. Wilks, M. Hamberg, and P.R. Ortiz de Montellano, "The Lipoxygenase Activity of Myoglobin," The Journal of Biological Chemistry (1994) Vol. 269, No. 10, pp. 7210-7216.
[36] M. Rizzo, G.A. Spinas, G.B. Rinia and K. Berneis, "Is diabetes the cost to pay for a greater cardiovascular prevention?" International Journal of Cardiology (2009), article in press; doi:10.1016/j.ijcard.2009.03.001
[37] A. Shahapurkar, S. M. Tarvade, N. M. Dedhia, S. Bichu, "Exertional Myoglobinuria Leading to Acute Renal Failure: A Case Report" Indian Journal of Nephrology (2004) Vol. 14, pp. 198-199.
[38] Y. Shimomura, M. Suzuki, S. Sugiyama, Y. Hanaki, and T. Ozawa, "Protective effect of coenzyme Q10 on exercise-induced muscular injury." Biochem Biophys Res Commun. (1991) Apr 15;176(1):349-55.
[39] J.P. Silva, M. Kohler, C. Graff, A. Oldfors, M.A. Magnuson, P.O. Berggren, and N.G. Larsson, "Impaired insulin secretion and beta-cell loss in tissue-specific knockout mice with mitochondrial diabetes" Nat Genet. (2000) Nov; Vol. 26, No. 3, pp. 336-40.
[40] H. Sinzinger, R. Wolfram, and B.A. Peskar, "Muscular side effects of statins," J Cardiovasc Pharmacol (2002) Vol. 40, pp. 163-71.
[41] R. Sukhija, MD, S.Prayaga, MD, M. Marashdeh, MD, Z. Bursac, PhD, MPH, P. Kakar, MD, D. Bansal MD, R. Sachdeva, MD, S.H. Kesan, MD, and J.L. Mehta, MD, PhD, "Effect of Statins on Fasting Plasma Glucose in Diabetic and Nondiabetic Patients" Journal of Investigative Medicine (2009) March; Vol. 57, Issue 3, pp. 495-499; doi: 10.231/JIM.0b013e318197ec8b
[42] A. Termana and U.T. Brunk "The Aging Myocardium: Roles of Mitochondrial Damage and Lysosomal Degradation," Heart, Lung and Circulation (2005) June, Vol. 14, Issue 2, pp. 107-114; doi:10.1016/j.hlc.2004.12.023
[43] G. Wainwright, L. Mascitelli, and M.R. Goldstein, "Cholesterol-lowering Therapy and Cell Membranes. Stable Plaque at the Expense of Unstable Membranes?" Arch. Med. Sci. (2009) Vol. 5, No. 3, pp. 289-295.
[44] T. Walker, J. McCaffery and C. Steinfort, "Potential link between HMG-CoA reductase inhibitor (statin) use and interstitial lung disease," MJA (2007) Vol. 186, No. 2, pp. 91-94.
[45] P.L. Yeagle, The Biology of Cholesterol (1988) 242 pp. CRC Press, Boca Raton, FL.
[46] F. Xia, L. Xie, A. Mihic, X. Gao, Y. Chen, H.Y. Gaisano and R.G. Tsushima, "Inhibition of Cholesterol Biosynthesis Impairs Insulin Secretion and Voltage-Gated Calcium Channel Function in Pancreatic Beta-Cells," Endocrinology (2008) Vol. 149, No. 10, pp. 5136-5145.
[47] R.A. Zager and K.M. Burkhart, "Differential effects of glutathione and cysteine on Fe2+, Fe3+, H2O2 and myoglobin-induced proximal tubular cell attack," Kidney Inernational (1998) Vol. 53, No 6, pp. 1661-1672. doi:10.1046/j.1523-1755.1998.00919.x
[2] M. Baciu, S.C. Sebai, O. Ces, X. Mulet, J.A. Clarke, G.C. Shearman, and R.V. Law, Templer RH, Plisson C, Parker CA, Gee A. "Degradative transport of cationic amphiphilic drugs across phospholipid bilayers." Philos Transact A Math Phys Eng Sci. (2006) Oct 15, Vol. 364(1847), pp. 2597-614.
[3] M.W. Bolt,J. W. Card, W.J. Racz, J.F. Brien and T.E. Massey, "Disruption of Mitochondrial Function and Cellular ATP Levels by Amiodarone and N-Desethylamiodarone in Initiation of Amiodarone-Induced Pulmonary Cytotoxicity," JPET (2001) September 1, Vol. 298, No. 3, pp. 1280-1289.
[4] S.L. Bonting, P.J. van Breugel, F.J. Daemen, "Influence of the lipid environment of the properties of rhodopsin in the photoreceptor membrane," Adv. Exp. Med. Biol. (1977) Vol. 83, pp. 175-89.
[5] P. Brancaccio, N. Maffulli, and F.M. Limongelli, "Creatine kinase monitoring in sport medicine" British Medical Bulletin (2007) Vol. 81-82. No. 1, pp. 209-230; doi:10.1093/bmb/ldm014
[6] E. Bruckert, G. Hayem, S. Dejager, et al. "Mild to moderate muscular symptoms with high-dosage statin therapy in hyperlipidemic patients -- the PRIMO study." Cardiovasc Drugs Ther (2005) Vol. 19, pp. 403-14.
[7] J. Cable, "Adverse Events of Statins -- An Informal Internet-based Study," JOIMR (2009, December, Vol. 7, No. 1; http://www.joimr.org/JOIMR_Vol7_No1_Dec2009.pdf.
[8] L.P. Cahalin, PT, PhD, et al., CHEST 2009: American College of Chest Physicians Annual Meeting, Poster 592. Presented November 4, 2009.
[9] J.C. Chatham, "Lactate - the forgotten fuel!" J Physiol. (2002) July 15; 542(Pt 2), p. 333. doi: 10.1113/jphysiol.2002.020974.
[10] E. Colman, A. Szarfman, J. Wyeth, et al., "An evaluation of a data mining signal for amyotrophic lateral sclerosis and statins detected in FDA"s spontaneous adverse event reporting system," Pharmacoepidemiol Drug Saf (2008) Vol. 17, pp. 1060-76.
[11] B.E. Corkey, J.T. Deeney, G.C. Yaney, K. Tornheim, and M. Prentki, "The Role of Long-Chain Fatty Acyl-CoA Esters in Beta-Cell Signal Transduction," American Society for Nutritional Sciences, (2000) pp. 299S-304S.
[12] S. Dublin, M.L. Jackson, J.C. Nelson, N.S. Weiss, E.B. Larson, and L.A. Jackson, "Statin use and risk of community acquired pneumonia in older people: population based case-control study," BMJ (2009) Vol. 338, p. b2137 ; doi:10.1136/bmj.b2137
[13] R.J. Edison and M. Muenke, "Central nervous system and limb anomalies in case reports of first-trimester statin exposure," N Engl J Med (2004) Vol. 350, pp. 1579-1582.
[14] J.L. Farber, "Mechanisms of cell injury by activated oxygen species." Environ Health Perspect. (1994) December; Vol. 102 (Suppl 10), pp. 17-24.
[15] A.B. Fernandez, R.H. Karas, A.A. Alsheikh-Ali, and P.D. Thompson, "Statins and interstitial lung disease: a systematic review of the literature and of food and drug administration adverse event reports." Chest, (2008) Oct, Vol. 134 No. 4, pp. 824-30. Epub 2008 Aug 8.
[16] K. Folkers, P. Langsjoen, R. Willis, P. Richardson,L.J. Xia,C.Q. Ye, and H. Tamagawa, "Lovastatin decreases coenzyme Q levels in humans," PNAS (1990) November 1, Vol. 87, No. 22, pp. 8931-8934.
[17] C. Gaugler, "Lipofuscin", Stanislaus Journal of Biochemical Reviews
May (1997).
[18] Ghirlanda G, Oradei A, Manto A, Lippa S, Uccioli L, Caputo S, Greco A, Littarru G (1993). "Evidence of plasma CoQ10-lowering effect by HMG-CoA reductase inhibitors: a double-blind, placebo-controlled study". J Clin Pharmacol 33 (3): 226-9. PMID 8463436.
[19] M.R. Goldstein, L. Mascitelli, and F. Pezzetta, "Dyslipidemia is a protective factor in amyotrophic lateral sclerosis," Neurology (2008) Vol. 71, p. 956.
[20] T. H. Haines, "Do Sterols Reduce Proton and Sodium Leaks through Lipid Bilayers?" Progress in Lipid Research (2001) Vol.40, pp. 299-324.
[21] S. Jamil and P. Iqbal, "Rhabdomyolysis induced by a single dose of a statin." Heart (2004) Jan; Vol. 90 No. 1, p. e3.
[22] N. Kucerka, D. Marquardt, T.A. Harroun, M-P Nieh, S. R. Wassall, and J. Katsaras, "The Functional Significance of Lipid Diversity: Orientation of Cholesterol in Bilayers is Determined by Lipid Species," J. Am. Chem. Soc. (2009) Vol. 131, pp. 16358-16359.
[23] J.M. Land, J.A. Morgan-Hughes, and J.B.Clark, " Mitochondrial myopathy: biochemical studies revealing a deficiency of NADH-cytochrome b reductase activity." J. Neurol. Sci. 50: 1-13, 1981.
[24] S. Lantuejoul, E. Brambilla, C. Brambilla, and G. Devouassoux, "Statin-induced Fibrotic Nonspecific Interstitial Pneumonia," Eur Respir J. (2002) Vol. 19, pp. 577-580.
[25] R.S. Lees and A.M. Lees, "Rhabdomyolysis from the Coadministration of Lovastatin and the Antifungal Agent Itraconazole," NEJM (1995) Vol. 333, pp. 664-665.
[26] S.R. Majumdar, F.A. McAlister, D.T. Eurich, R.S. Padwal, and T.J. Marrie, "Statins and outcomes in patients admitted to hospital with community acquired pneumonia: population based prospective cohort study," BMJ (2006) Vol. 333, p. 999.
[27] M.G. Mohaupt, MD, R.H. Karas, MD PhD, E.B. Babiychuk, PhD, V. Sanchez-Freire, K. Monastyrskaya, PhD, L. Iyer, PhD, H. Hoppeler, MD, F. Breil and A. Draeger, MD "Association between statin-associated myopathy and skeletal muscle damage," CMAJ (2009) July 7 Vol. 181 No. 1-2 ; doi:10.1503/cmaj.081785.
[28] A. Mordente, S. A. Santini, G. A. D. Miggiano, G. E. Martorana, T. Petitti, G. Minotti, and B. Giardina, "The Interaction of Short Chain Coenzyme Q analogs with Different Redox States of Myoglobin," The Journal of Biological Chemistry, (1994) Vol. 269, Mo. 44, pp. 27394-27400.
[29] R.A. Oleka, J. Antosiewicza, J. Popinigisa, R. Gabbianellib, D. Fedelib and G. Falcionib, "Pyruvate but not lactate prevents NADH-induced myoglobin oxidation," Free Radical Biology and Medicine (2005) June; Vol. 38, Issue 11, pp. 1484-1490; doi:10.1016/j.freeradbiomed.2005.02.018.
[30] D. Papahadjopoulosa "Na+-K+ discrimination by “pure” phospholipid membranes," Biochimica et Biophysica Acta (BBA) (1971) Vol. 241, Issue 1, 6 July 1971, pp. 254-259
[31] R.P. Patel, U. Diczfalusy, S. Dzeletovic, M.T. Wilson and V.M. Darley-Usmar, "Formation of oxysterols during oxidation of low density lipoprotein by peroxynitrite, myoglobin, and copper, "Journal of Lipid Research (1996) Vol. 37, pp. 2361-2371.
[32] S. Pitkanen, A. Feigenbaum,, R. Laframboise, and B.H. Robinson, "NADH-coenzyme Q reductase (complex I) deficiency: heterogeneity in phenotype and biochemical findings," J. Inherit. Metab. Dis. (1996) Vol. 19, pp. 675-686.
[33] E.Y. Plotnikov, A.A. Chupyrkina, I.B. Pevzner, N.K. Isaev, and D.B. Zorov, "Myoglobin causes oxidative stress, increase of NO production and dysfunction of kidney's mitochondria," Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease (2009) Vol. 1792, Issue 8, August pp. 796-803; doi:10.1016/j.bbadis.2009.06.005
[34] T. Pozefsky, R G Tancredi, R T Moxley, J Dupre, and J D Tobin "Effects of brief starvation on muscle amino acid metabolism in nonobese man." J Clin Invest. (1976) February, Vol. 57, No. 2, pp. 444-449. doi: 10.1172/JCI108295.
[35] S.I. Rao, A. Wilks, M. Hamberg, and P.R. Ortiz de Montellano, "The Lipoxygenase Activity of Myoglobin," The Journal of Biological Chemistry (1994) Vol. 269, No. 10, pp. 7210-7216.
[36] M. Rizzo, G.A. Spinas, G.B. Rinia and K. Berneis, "Is diabetes the cost to pay for a greater cardiovascular prevention?" International Journal of Cardiology (2009), article in press; doi:10.1016/j.ijcard.2009.03.001
[37] A. Shahapurkar, S. M. Tarvade, N. M. Dedhia, S. Bichu, "Exertional Myoglobinuria Leading to Acute Renal Failure: A Case Report" Indian Journal of Nephrology (2004) Vol. 14, pp. 198-199.
[38] Y. Shimomura, M. Suzuki, S. Sugiyama, Y. Hanaki, and T. Ozawa, "Protective effect of coenzyme Q10 on exercise-induced muscular injury." Biochem Biophys Res Commun. (1991) Apr 15;176(1):349-55.
[39] J.P. Silva, M. Kohler, C. Graff, A. Oldfors, M.A. Magnuson, P.O. Berggren, and N.G. Larsson, "Impaired insulin secretion and beta-cell loss in tissue-specific knockout mice with mitochondrial diabetes" Nat Genet. (2000) Nov; Vol. 26, No. 3, pp. 336-40.
[40] H. Sinzinger, R. Wolfram, and B.A. Peskar, "Muscular side effects of statins," J Cardiovasc Pharmacol (2002) Vol. 40, pp. 163-71.
[41] R. Sukhija, MD, S.Prayaga, MD, M. Marashdeh, MD, Z. Bursac, PhD, MPH, P. Kakar, MD, D. Bansal MD, R. Sachdeva, MD, S.H. Kesan, MD, and J.L. Mehta, MD, PhD, "Effect of Statins on Fasting Plasma Glucose in Diabetic and Nondiabetic Patients" Journal of Investigative Medicine (2009) March; Vol. 57, Issue 3, pp. 495-499; doi: 10.231/JIM.0b013e318197ec8b
[42] A. Termana and U.T. Brunk "The Aging Myocardium: Roles of Mitochondrial Damage and Lysosomal Degradation," Heart, Lung and Circulation (2005) June, Vol. 14, Issue 2, pp. 107-114; doi:10.1016/j.hlc.2004.12.023
[43] G. Wainwright, L. Mascitelli, and M.R. Goldstein, "Cholesterol-lowering Therapy and Cell Membranes. Stable Plaque at the Expense of Unstable Membranes?" Arch. Med. Sci. (2009) Vol. 5, No. 3, pp. 289-295.
[44] T. Walker, J. McCaffery and C. Steinfort, "Potential link between HMG-CoA reductase inhibitor (statin) use and interstitial lung disease," MJA (2007) Vol. 186, No. 2, pp. 91-94.
[45] P.L. Yeagle, The Biology of Cholesterol (1988) 242 pp. CRC Press, Boca Raton, FL.
[46] F. Xia, L. Xie, A. Mihic, X. Gao, Y. Chen, H.Y. Gaisano and R.G. Tsushima, "Inhibition of Cholesterol Biosynthesis Impairs Insulin Secretion and Voltage-Gated Calcium Channel Function in Pancreatic Beta-Cells," Endocrinology (2008) Vol. 149, No. 10, pp. 5136-5145.
[47] R.A. Zager and K.M. Burkhart, "Differential effects of glutathione and cysteine on Fe2+, Fe3+, H2O2 and myoglobin-induced proximal tubular cell attack," Kidney Inernational (1998) Vol. 53, No 6, pp. 1661-1672. doi:10.1046/j.1523-1755.1998.00919.x
Subscribe to:
Posts (Atom)